一種基於分子動力學模擬篩分黃酮類化合物抗雄活性方法與流程
2023-09-21 09:30:35 6
本發明屬於生物領域,特別涉及一種通過分子動力學模擬方法來考察黃酮類化合物與雄激素受體的構型變化而篩分黃酮類化合物的抗雄活性的方法。
背景技術:
內分泌幹擾物是指可模擬人體激素而擾亂人體內分泌幹擾系統的物質,不僅影響生物的生殖系統、誘發癌症,甚至會禍及第二代、第三代。雌激素受體、雄激素受體和甲狀腺受體作為核受體超家族的重要組成部分,是介導內分泌幹擾效應的重要靶點。一部分內分泌幹擾物可以模擬天然激素激活這些靶點,該效應稱為擬-雌/雄/甲狀腺激素效應,另一部分內分泌幹擾物可以抑制天然激素的作用,這一效應稱為抗-雌/雄/甲狀腺激素效應。目前已被報導的相關的內分泌幹擾物種類繁多,比如雙酚A類、酞酸酯類、多氯聯苯類、多溴聯苯醚類等等。不僅如此,每年新增的化學品更是不計其數,需要對海量化學品的潛在內分泌幹擾活性進行高效率篩分。針對這三類受體介導的內分泌幹擾物,已經建立了許多實驗方法用以篩選。目前主要的篩選方法有基於體內的生物標誌法、組學方法,體外試驗中的酵母雙雜交、報告基因實驗法以及競爭結合實驗等等。但是這些實驗方法費時費力,如果完全依靠它們來進行內分泌幹擾物的篩查是不現實的。
基於這樣一些挑戰,計算機虛擬篩選內分泌幹擾物成為了有力的工具和輔助手段,比如定量結構效應關係(quantitative structure-activity relationship,QSAR)已經被廣泛使用,並且取得了很多成果。QSAR方法的基礎是通過比較化合物的結構從而預測其活性,但現實中有大量的化合物結構相似,卻存在活性有無的差別。也就是說,目前的QSAR方法基於結構的相似活性相似的原理僅能夠預測那些已知有活性、且結構相似的化合物的活性,而不能夠判斷化合物活性有無。因此,在具體實踐過程中,絕大多數研究者們只選擇有活性化合物作為研究對象,經常忽視那些無活性化合物的存在,這是QSAR方法最大的缺陷,也是QSAR無法在預測化合物內分泌幹擾活性領域得到有效應用的真正原因。為了彌補這一缺陷,必須找到一種能預判化合物活性有無的快速有效方法。
大量研究表明,化合物產生內分泌幹擾效應主要通過以下幾個步驟:首先,化合物要與受體相互結合(發生在配體結合域)並相互作用,達到一個穩定狀態;其次,配體-受體複合物進行同源或者異源二聚化或四聚化;最後,二聚體或四聚體識別特定的染色體DNA序列,並與共調節因子(包括共激活和共抑制因子)結合,從而產生對目標基因的調控,即產生內分泌幹擾效應。配體與本發明涉及的雄激素受體結合發生在配體結合域中,該區域有11個α螺旋構成(為了與其他核受體名稱上的統一,把第二條螺旋直接命名為H3,第三條螺旋直接命名為H4,依次類推)。研究發現,該區域的構型變化將直接影響核受體的生物功能,其中尤為重要的是H12的位置的變化。一般認為,配體(也即小分子化合物)進入受體的配體結合域,與結合口袋內的殘基發生範德華作用、電相互作用、氫鍵作用、疏水作用等,在這些力的共同作用下,配體和受體發生運動,最終穩定在特定位置後,才能發生轉錄作用。對於受體而言,運動的主要特徵是其H12的變化,最終穩定在特定的位置,與其他相對穩定的螺旋提供一個與共激活因子/共抑制因子結合的表面。對於配體而言,其運動的主要特徵是是否能一直存在於結合區域,並且是否能在結合區域穩定存在。此外,由於雄激素受體屬於核受體家族中「類雌激素受體」,與甲狀腺激素受體(Thyroid hormone receptor,TR)等不同,該受體只發生同源二聚化,不會受到其他核受體轉錄的影響。因此,我們可以通過模擬手段考察配體、H12的運動來預判在真實的生物體系中,對應小分子的受體活性。
分子動力學模擬(molecular dynamics simulation,MD)是一種基於計算機考察原子和分子物理運動的模擬技術。其基本原理是,給定模擬體系的初始態,依靠牛頓力學來模擬分子體系的運動,以在由分子體系的不同狀態構成的系綜中抽取樣本,從而計算體系的構型積分,並以構型積分的結果為基礎進一步計算體系的宏觀性質。該方法已經被大量用於解釋藥物激活靶點的機理上,但是作為一種準確篩分工具,卻從來沒有被應用。該技術可以作為QSAR預測化合物內分泌幹擾效應的前提技術。
張愛茜等曾將分子動力學技術用於識別具有ER激動和拮抗作用的有機物(張愛茜,藺遠,彭素芬,劉磊,高常安,韓朔睽.一種有機物雌激素受體激動和拮抗作用的識別方法[P].江蘇:CN101381894,2009-03-11),但是該方法僅針對ER,且未引入蛋白變構的概念。
於紅霞等曾將分子動力學技術用於篩選核受體介導內分泌幹擾物(於紅霞,史薇,王小享.基於分子動力學模擬的核受體介導內分泌幹擾物質的虛擬篩選方法[P].江蘇:CN201310288617.3,213-9-25),但該方法僅能通過比對波形進行大致判斷,無法進行定量預測,預測精度低,實用性差。
技術實現要素:
本發明的目的是根據雄激素受體與黃酮類化合物之間的相互作用關係,提供一種基於計算機技術、省時省力經濟的篩分黃酮類化合物抗雄活性方法,為化學品安全和風險評估提供技術支撐。
為了實現上述目的,本發明採用以下技術方案:一種基於分子動力學模擬篩分黃酮類化合物抗雄活性方法,包括以下步驟:
(1)構建黃酮類化合物和雄激素受體的配體-受體複合物分子結構;
(2)對分子配體-受體複合物分子結構進行動力學模擬;
(3)建立模擬結果區分模型;
(4)對模擬結果根據模型進行判斷:如果化合物位於有活性區域,則判斷化合物為有抗雄活性;如果化合物位於無活性區域,則判斷化合物為無抗雄活性;如果化合物位於模糊區域,則進行步驟五和六;
(5)模擬結果中配體在A環有甲氧基且受體於配體形成的氫鍵數量少於設定值時,判斷判斷化合物為無抗雄活性;
(6)觀察受體和配體運動軌跡,如果配體的運動軌跡已經脫離了雄激素受體的結合口袋,判定化合物無抗雄活性;如果受體在模擬過程結束後出現「mousetrap」現象,判定化合物有抗雄活性;
(7)如化合物經步驟五和六均判斷為無抗雄活性,則判斷化合物為無抗雄活性,否則判斷化合物為有抗雄活性;
(8)對判斷有抗雄活性的化合物建立CoMSIA預測模型,預測具有抗雄活性黃酮化合物的活性大小。
所述步驟1中配體-受體複合物分子結構的構建採用以下步驟:構建黃酮類化合物的分子結構和雄激素受體初始態結構,進行能量優化,並賦予Gasteiger-Huckel電荷;利用Sybyl7.3的軟體將配體對接入受體文件,範圍閾值和膨脹係數分別為0.5和0,選取對接得分最高的構象作為初始構象,將之與受體合併,組成複合物;
所述雄激素受體的初始態結構採用雄激素受體的胺基酸序列文件,導入Swiss-Model網絡伺服器中,然後以ERα初始態的蛋白晶體結構為模板進行同源模建。
所述步驟2中進行動力學模擬,採用GROMACS軟體進行,對配體和受體都賦予CHARMM27力場,其中受體的力場參數為軟體自帶,而配體的力場參數由Swiss-Param賦予;將整個系統放入被TIP3P水分子佔據的模型立方體盒子中,複合物距離盒子邊界的最短距離為1納米,加入鈉離子或氯離子保證系統的電性平衡;用最陡下降法和共軛梯度法實現整個系統的能量收斂,然後系統升溫至300K並一直維持,並賦予1個標準大氣壓;約束受體蛋白後進行50ps的分子動力學模擬,最後撤銷約束進行模擬。
所述動力學模擬中電相互作用使用particle mesh Ewald算法,範德華力以1納米截斷半徑,模擬的步長2fs,一個體系進行20ns的模擬。
所述模擬結果區分模型建立採用以下步驟:選取48種典型的黃酮類作為篩選集,構建48個黃酮-雄激素受體複合物,體系經過預處理後使用Gromacs進行分子動力學模擬,體系維持在1atm,截斷半徑設為動力學模擬步長為2fs,每2ps保存一個構象,每個體系進行20ns的動力學模擬;然後對H12進行均方根偏差分析,有抗雄活性的化合物的H12在8ns保持穩定,而其他體系的H12一直保持波動;將H12在8~20ns的均方根偏差導入R語言,獲得區分模型。
本發明的基本原理是利用分子動力學計算雄激素受體結合受試化合物後發生的構型變化,基於H12鏈在8~20ns時間波動情況、化合物與受體在8~20ns時間內形成氫鍵情況、配體小分子運動軌跡、配體小分子在模擬期間波動情況來綜合判定受試化合物是否有相關的受體活性。
採用本發明方法可以模擬黃酮類化合物於雄激素受體結合後相互作用的動態過程,基於分子動力學模擬鑑別物質是否具有受體抗性,然後利用CoMSIA模型來預測這些有抗性但活性未知的黃酮類化合物活性。該方法成本低廉,操作簡便,為大規模預測黃酮類化合物抗雄活性提供了用力的工具,也為進一步篩分具有抗雄活性的黃酮類藥物提供支撐。
本方法首次引入定量判斷,並運用定量和定性結合來提高預判的精確度,使得抗性和無抗性黃酮的區分率達到87.5%,無抗性黃酮的篩除率達到90%,不僅能夠大幅削減實驗室工作量,減少試驗品消耗,節約資源,還能大幅提高QSAR預測的準確性,使得QSAR能夠真正實現應用。
附圖說明
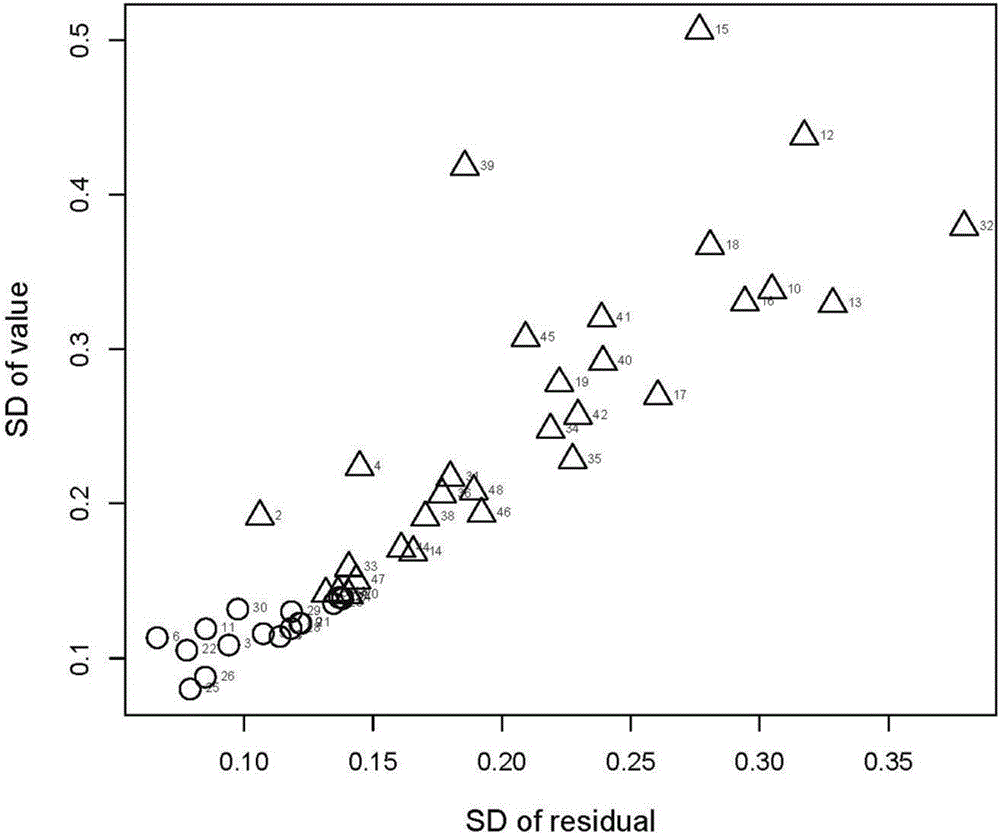
圖1是實施例的區分模型,圓圈代表實測有活性化合物,三角形代表實測無活性化合物。
具體實施方式
以下通過實施例進一步說明本發明,但本發明的保護範圍不僅限於實施例。
(1)在chemdraw內構建受試化合物的分子結構,在高斯09軟體中用密度泛函方法b3lyp/6-311G*基組進行能量優化,然後利用Sybyl7.3軟體賦予Gasteiger-Huckel電荷。本研究中需要的是受體的自由態結構,從蛋白質資料庫(PDB)獲得ER的自由態結構,其PDB編號是1A52,然後以1A52作為模板,利用Swiss-Model網絡伺服器構建AR的初始結構。構建的結果用拉氏圖法進行檢驗,證明結果的合理性。所有結構都用spdv檢查,缺失的殘基用該軟體自動補齊。然後利用Sybyl7.3軟體的Surflex-dock模塊尋找受體大分子活性口,將受試化合物分別對接入口袋。選取對接打分值最高的化合物構象作為活性構象,將之與受體合併,組成複合物用於分子動力學模擬。
(2)然後對構建好的配體-受體複合物進行分子動力學模擬。的分子動力學模擬都是基於GROMACS軟體包,該軟體相比於AMBER等其他分子動力學軟體具有更快速的運算速度,適宜於大規模的篩選。對配體和受體都賦予CHARMM27力場,其中受體的力場參數為軟體自帶,而配體的力場參數有Swiss-Param賦予。將整個系統放入被TIP3P水分子佔據的模型立方體盒子中,複合物距離盒子邊界的最短距離為1納米。為了保證系統的電性平衡,在體系中加入鈉離子或氯離子。用最陡下降法和共軛梯度法實現整個系統的能量收斂,然後系統升溫至300K並一直維持,並賦予1個標準大氣壓。約束受體蛋白後進行50ps的分子動力學模擬,最後撤銷約束進行模擬。電相互作用使用particle mesh Ewald(PME)算法,範德華力以1納米截斷半徑,模擬的步長2fs,一個體系一般進行20ns的模擬。
(3)根據模擬的結果提取受體H12(receptor-H12)空間位置的均方根偏差(root mean square deviation,RMSD),使用R語言包計算其標準偏差和標準殘差:RMSD(i)=a×time(i)+b+ε(i),其中ε是殘差,a和b是常數。建立標準偏差和殘差之間的相關性,獲得區分模型。當化合物位於有活性區域時,認為該化合物為有抗雄活性;但化合物位於無活性區域時,認為該化合物無抗雄活性;當化合物位於模糊區域時,則需要進行進一步判斷。
(4)根據模擬結果提取受體-配體相互作用過程中形成的氫鍵(hydrogen bond),分析8-20ns間受體於配體形成的氫鍵數量。當配體在A環有甲氧基且在8-20ns間形成氫鍵數量很少,認為該化合物無抗雄活性。
(5)觀察受體和配體運動軌跡,如果配體的運動軌跡已經脫離了雄激素受體的結合口袋,判定化合物無抗雄活性;如果受體在模擬過程結束後出現「mousetrap」現象,判定化合物有抗雄活性。
(6)使用技術方案(4)-(5)對技術方案(3)中無活性組合模糊組的化合物進行驗證和區分:若這兩組中化合物滿足技術方案(4)和(5)中關於有(無)活性判定,則化合物有(無)活性。
(7)運用sybyl軟體中CoMSIA模塊,基於已知黃酮化合物抗雄活性,建立CoMSIA預測模型,預測未知活性但判定具有抗雄活性黃酮化合物的活性大小。
實施例
初始態受體文件構建:
AR屬於「雌激素類」核受體,其初始態結構並未獲得,但ERα初始態的蛋白晶體結構已經由前人通過實驗獲得(PDB code:1A52)。「雌激素類」核受體的特點是在激活態/抑制態的H12位置差異明顯,因此AR也具有相同特性。從美國國家生物信息中心資料庫(national center for biotechnology information,NCBI)獲AR的胺基酸序列文件,導入Swiss-Model網絡伺服器中,然後以1A52為模板進行同源模建。同源模建完成後要在Pymol軟體中進行疊合處理,將新構建H11-H12以H11為疊合參考系疊合至實驗PDB文件,將構建的H12合併至原文件的H1-H11獲得新的結構文件,也即受體的初始態結構。過程中,需要的AR的PDB文件是1T7T。
配體-受體複合物構建:
利用本發明對蛋白文件和小分子文件進行預處理,用於進一步的分子對接與分子動力學模擬。在chemdraw12.0內繪製小分子的二維結構式,然後用chem3D12.0的MM2模塊進行初步優化。優化得到的結構用高斯軟體進行量子優化。其中量子優化利用密度泛函方法進行,在6-311G*基組上進行。最後對所有小分子統一地賦予Gasteiger-Huckel電荷。所有用於分子動力學模擬的受體文件也都要進行預處理。先用spdbv對胺基酸缺損的支鏈進行修補,在Sybyl7.3內對每個蛋白文件加氫原子,同時賦予Gasteiger-Huckel電荷。
利用Sybyl7.3的Surflex-Dock模塊將小分子對接入受體文件。用自動搜索方法搜尋對接口袋,範圍閾值和膨脹係數分別設為0.5和0。然後將小分子對接入對接口袋,每一個小分子將得到得分最高的是個構象,我們選取得分最高的構象作為分子動力學模擬的初始構象。
建立黃酮類的抗雄激素活性區分模型:
選取48種典型的黃酮類作為篩選集,採用本發明構建48個黃酮-AR複合物,體系經過預處理後使用Gromacs進行分子動力學模擬。其中包括結構10000步最陡下降法優化、50ps升溫至300K、200ps的常溫常壓(NPT)系綜平衡和最終的模擬過程。在模擬過程中,體系維持在1atm,截斷半徑設為動力學模擬步長為2fs,每2ps保存一個構象用於結果分析。
每個體系進行20ns的動力學模擬,然後通過對H12的RMSD的分析發現,有抗雄活性的化合物的H12在8ns之前就保持穩定,而其他體系的H12一直保持波動。我們將H12在8-20ns的RMSD導入R語言,獲得區分模型,13個黃酮化合物位於有活性區,27個位於無活性區、7個位於模糊區。分析黃酮化合物8-20ns模擬時間內生成的氫鍵數量,當某一化合物在A環有甲氧基但很少生成氫鍵的情況下,認為該化合物無活性。在活性區域中的櫻黃素(prunetin)由於在A環有甲氧基且很少氫鍵,認為該化合物無抗雄活性;在模糊區域中的7-甲氧基黃酮也同樣為無抗雄活性。此外,在活性組中,6-甲氧基黃烷酮表現出「mousetrap」現象,再一次確定活性;在非活性組中,3-羥基-6-甲氧基黃酮直接在模擬過程中從結合口袋中逃逸,再次確定無活性。通過本發明,48個黃酮化合物中的42個可以判定是否具有抗雄活性;31個無抗雄活性黃酮化合物中的27個可以被明確鑑定出來,鑑別率高達90%。鑑別結果與我們通過MDA-kb2實驗結果和文獻結果相一致。
以上所述,僅是本發明的較佳實施例而已,並非對本發明做任何形式的限制。凡是依據本發明的技術和方法實質對以上實施例所作的任何簡單修改、等同變化與修飾,均仍屬於本發明的技術和方法方案的範圍內。