使用親和樹脂合成ADC的方法與流程
2023-09-16 12:25:45 4
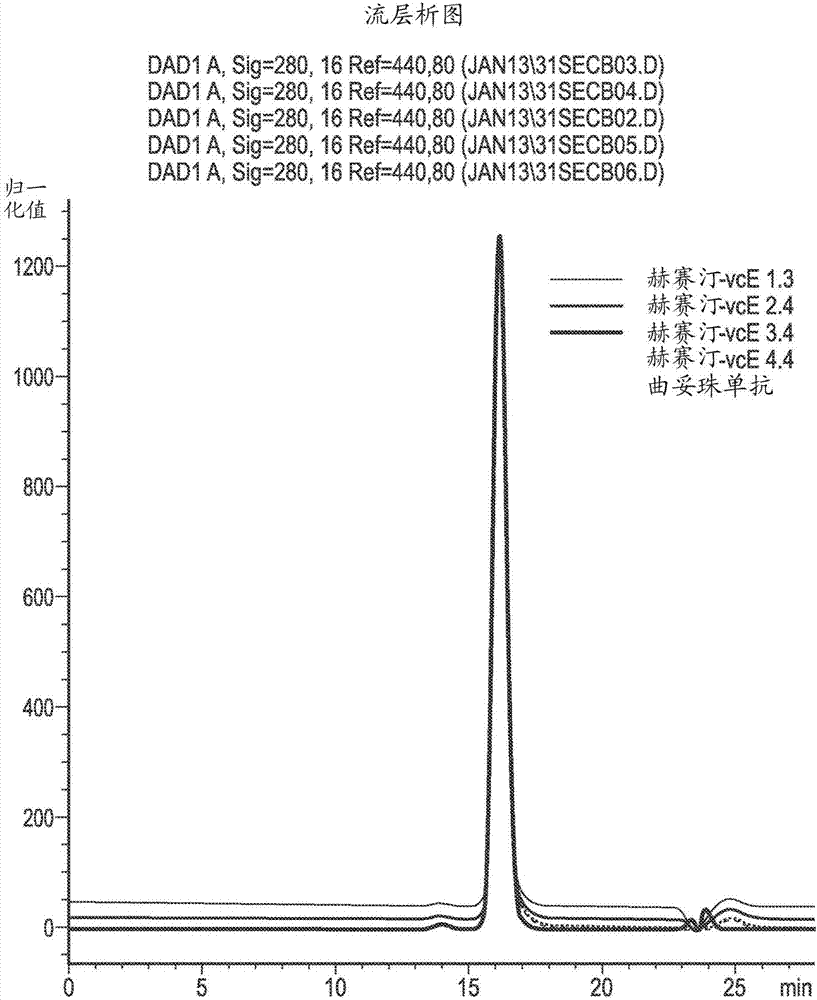
本發明涉及合成生物分子-藥物-綴合物(biomolecule-drug-conjugates)的固相方法。特別地,本發明涉及合成抗體-藥物-綴合物(adc)的固相方法。本發明還涉及產生固定化的、化學修飾的生物分子,例如抗體的中間方法。除了以上方法以外,本發明涉及本發明的方法的捕獲樹脂、生物分子-藥物-綴合物、中間產物和組合物的多種用途。背景免疫毒素和抗體藥物綴合物(adc)為將靶特異性結合結構域與可以被歸類為細胞毒性的具有足夠有效的毒性的藥物分子結合的蛋白質藥物。抗體為用於創建將高特異性與高抗原親和性結合、允許細胞毒性藥物向期望施用的位點轉運的靶向系統的該目的的理想生物分子。這些藥物構建體針對疾病為潛在地治療性的,在腫瘤學中特別流行。抗體藥物綴合物(adc)的主要標準為毒素『彈頭』(藥物)以極低水平(pm)具有活性。此外,針對腫瘤細胞具有療效而不考慮周期中的點為有利的。出於該目的,發現dna活性劑作為毒素候選物為有利的,因為dna損傷(除非可修復)將驅動細胞凋亡而不考慮周期中的點。原則上,adc的合適的細胞毒性或細胞抑制性藥物有效負載(payload)可以為被定義為以下的任何部分:l01atc分子(『解剖學治療學化學分類系統(anatomicaltherapeuticchemicalclassificationsystem)』,其中l01為定義抗腫瘤的和免疫調節的劑的亞組,由who藥物統計方法合作中心(whocollaboratingcentrefordrugstatisticsmethodology)定義)。可選擇地,可以被歸類為adc的合適的有效負載的其他部分可以被簡單地定義為被內化後對細胞有毒的任何物質。落入後一類別的大多數部分將缺乏足夠有效的效力。因此,存在鑑定和利用『超效力(ultra-potency)』物質的行業趨勢(industrytrends)。在撰寫時,目前在臨床試驗中存在>33種adc,並在早期評價中還存在>250種adc。關於免疫毒素和adc研究的基本原理、設計和有效性的專家綜述可以見於以下中:j.adair等人,expertopin.biol.ther.,2012,12(9):p1191-206,g.casi等人,journalofcontrolledrelease,2012,161,2,p422–428,f.dosio等人,toxins,2011,3,p848-883和s.panowski等人mabs2014,6:1,34–45。許多溶液相方法可以用於製備生物分子-藥物-綴合物,例如抗體-藥物-綴合物(adc)。然而,溶液相方法本身在生成大體積廢棄物方面為浪費的,並且在合成期間在生物分子-藥物-綴合物的聚集方面為有問題的。用於製備生物分子-藥物-綴合物的溶液相方法中的第一步通常涉及生物分子的化學修飾或活化。例如,在生物分子為抗體的情況下,抗體可以通過還原或部分還原抗體而被『化學修飾』或『活化』。用於部分還原抗體的合適的過程在「bioconjugatetechniques」,第96/97頁,gregt.hermanson,academicpress;第二版,2008,isbn-13:978-0123705013中給出。在還原過程中通常採用還原劑諸如tcep。在抗體的化學修飾或活化例如還原之後,下一步通常為去除任何過量的活化/化學修飾劑,例如過量的還原劑。該步驟非常耗時,因為通常需要將樣品通過分離柱運行多次。如果生物分子的穩定性為問題,這可在降解方面也為有問題的。可選擇地可以應用滲濾步驟,但這可以導致在處理期間的物質的損失。在以上純化步驟之後,然後將化學修飾的/活化的例如還原的抗體與藥物部分綴合。伴隨該步驟的主要問題為生物分子-藥物-綴合物聚集的高概率。當在該過程中採用高度疏水藥物有效負載時,這為特別有問題的。聚集為主要問題,因為它可以導致不可用的生物分子-藥物-綴合物。在最好的情況下,被生物分子-藥物-綴合物聚集體汙染的生物分子-藥物-綴合物必須進一步被純化以去除聚集體,這既耗時又非常浪費。在純化期間大部分藥物將被損失,因為它形成聚集的生物分子-藥物-綴合物的部分。在最壞的情況下,整批生物分子-藥物-綴合物被生物分子-藥物-綴合物聚集體汙染至如此高的程度以至於其完全不可用並必須被處置。生物分子-藥物-綴合物的純化步驟可以非常耗時,因為通常需要將綴合物通過柱運行多次。腫瘤學家已經致力於利用靶特異性單克隆抗體將細胞毒性藥物遞送至腫瘤部位(只要單克隆抗體存在)近三十年了。直到現在,三類毒素已經在該領域佔首要地位。即,加利車黴素(calicheamicins)、美登素(maytansines)和奧瑞他汀(auristatins)。這些細胞毒性藥物種類本質上通常都為疏水的。當與抗體綴合時,它們的存在顯著增加了抗體的總體疏水性,並且在一些情況下達到綴合物之間的疏水相互作用導致綴合物聚集的程度(y.adem等人,bioconjugatechem.2014,25,656-664)。基於麥羅塔(mylotarg)和cmc-544兩者的過程均包含層析聚集體去除步驟的認識,該問題的顯著性的順序為加利車黴素>美登素>奧瑞他汀。約50%的美登素過程包含聚集體去除步驟,並且非常少的奧瑞他汀過程包含聚集體去除步驟。最近,基於倍癌黴素(duocarmycin)(www.syntarga.com)、吡咯並苯二氮卓類(pyrrolobenzodiazepine)(pbd)二聚體(www.spirogen.com)和α鵝膏毒環肽(www.heidelberg-pharma.com)的細胞毒性毒素有效負載已經與抗體綴合,並且正在經歷臨床前評價。這些新種類的毒素比其前任(predecessor)細胞毒素藥物種類更疏水,並且當經由隨機工具(means)與抗體綴合時,更易於聚集(s.jeffrey等人,bioconjugatechem.2013,24,1256-1263)。聚集為物理不穩定性的原因,並且可以為抗體綴合產物諸如adc的限制性穩定性參數。因為這些物質對患者具有重要的療效和毒性作用,所以聚集體含量應被保持在最低限度(m.manning等人,pharm.res;2010,27,544-75)。很大的努力集中於通過摻入親水接頭來調節藥物的疏水性(zhao等人,j.med.chem.,2011,54,10,3606–3623)。在不能控制聚集體形成的情況下,開發人員已經依賴於熟知的技術用於從基於蛋白的治療劑(therapeutics)去除聚集體。這些包括一系列不同的層析分離,包括離子交換、疏水相互作用、羥基磷灰石(wo03/057163)和本領域技術人員熟知的其他層析分離(a.wakankar等人mabs,2011mar-apr;3,2,161)。最近發現切向流過濾純化為有利的(wo2006086733)。進行此類層析純化技術具有實現足夠產品質量的結果,但具有挑戰性並且通常以過程產量為代價。當在製備的場景下操作抗體和基於抗體的治療劑時,通過不明確的、偶然的副反應的物質的物理損失或通過聚集體形成的不想要的生理化學相互作用具有非常大的顯著的財務影響。manning等人(pharm.res.,2010,27,4,544)將聚集體定義為(i)快速可逆的非共價的小的寡聚體(二聚體、三聚體、四聚體等);(ii)不可逆的非共價的寡聚體;(iii)共價的寡聚體(例如,二硫鍵連接的);(iv)大的聚集體(>10mer’s),如果為非共價的其可以為可逆的;(v)非常大的聚集體(直徑為50nm至3000nm),如果為非共價的其可以為可逆的;和(vi)可視顆粒(『雪』),其可能為不可逆的。聚集可以由非共價相互作用引起或由共價連接的物類(species)引起。當比較單體抗體綴合物的生物活性與相應聚集體的生物活性時,這些高分子量物類的存在可以顯著損害綴合物的效力。在此類情況下,產品療效可能受損(m.vazquez-rey等人,biotechnologyandbioengineering,2011,第108卷,7,1495)。聚集體形成對生物分子或抗體綴合物中的單體純度具有直接和負面影響。聚集為主要問題,因為它可以導致不可用的生物分子和抗體綴合物。在最壞的情況下,整批綴合物將被聚集體汙染至如此高的程度以至於其完全不可用且不適於多次純化,並且因此必須被處置。應當理解,抗體藥物綴合物中的聚集程度與摻入抗體的疏水藥物毒素的程度成正比。對於其中抗體用細胞毒性有效負載衍生化的隨機綴合,所得綴合物將包括一定展寬(aspread)的藥物抗體比(drugantibodyratio)(dar)物類。當靶向4的dar時,dar物類的展寬將為高斯的(gaussian)並且通常在0和8之間。對於易於聚集的綴合物,這些較高dar物類將通常存在於聚集體中。在製備中,這種現象具有減少總體綴合物的dar、引起該過程遠離靶dar特化(specification)的作用。因此,聚集對實現抗體綴合物的靶dar具有負面影響。對adc世界會議的參與者進行了調查,以了解他們認為什麼是adc發展最大的cmc障礙。超過一半的參與者(52%)認為不想要的聚集為主要問題。40%的參與者還強調了對控制dar的關注,其中20%警告從綴合物產品物質中去除多餘的未結合藥物(antibody-drugconjugatesindustryoutlook2014「insightandanalysisofthemajortrends,challengesandopportunitieswithintheadcfield」hansonwade,第10頁)。fda對adc均勻性的要求越來越多,鼓勵開發人員證明綴合事件的精確位置。為了解決這些要求,目前最先進的抗體綴合技術實現了位點特異性綴合技術,其中綴合事件僅在工程化半胱氨酸殘基(thiomab,genentech)、非天然胺基酸(azido,synafix)或經由酶促工具(smartag,redwoodbioscience)經由抗體上的特定位置上的工程化標誌發生。通過控制藥物抗體比(dar),位點特異性綴合導致均一性更高的adc產生。改進dar對綴合物穩定性、體內耐受性、藥代動力學(pk)性質和療效有正面影響。通常,位點特異性綴合技術靶向低dar,通常dar為2。為了實現療效並實現治療窗口,細胞毒性有效負載必須具有精準效力,因為每抗體的綴合事件的數目為有限的。通常有此類精準效力的細胞毒性有效負載在本質上為高度疏水的,並且因此易於發生聚集效應。儘管位點特異性綴合有這些優點,聚集問題仍然存在。對於具有聚集傾向的所有抗體綴合技術,綴合過程以相對低的濃度(通常約1g/l)進行。稀溶液相方法的使用為嘗試規避聚集效應的直接方法。然而,即使在此類濃度下,仍然可以觀察到聚集效應,並且其不利於產品質量,需要純化整個過程批次。用於製備的稀釋溶液和用於純化的另外的過程介質的使用積累大量的必須作為細胞毒性廢棄物處理的廢棄溶劑。因此,避免此類廢棄物積累的製備過程對環境更有利和高效。因此,用於製備生物分子-藥物-綴合物的常規溶液相方法困難重重,並且將期望提供製備生物分子-藥物-綴合物的改進方法。本發明解決了常規的溶液相方法的一個或更多個以上問題。公開內容概述合成生物分子-藥物-綴合物的方法:根據本發明,提供了一種合成生物分子-藥物-綴合物的方法,該方法包括:a)任選地使生物分子與化學修飾劑、酶促修飾劑或活化劑接觸,以提供化學修飾的、酶促修飾的或活化的生物分子;b)(i)當進行步驟(a)時,使步驟(a)的化學修飾的、酶促修飾的或活化的生物分子與包含基於非肽的蛋白a、蛋白g或蛋白l模擬物生物分子捕獲部分的捕獲樹脂在適於固定化化學修飾的、酶促修飾的或活化的生物分子的條件下接觸,並因此提供固定化的化學修飾的、酶促修飾的或活化的生物分子;或(ii)當不進行步驟(a)時,使生物分子與包含基於非肽的蛋白a、蛋白g或蛋白l模擬物生物分子捕獲部分的捕獲樹脂在適於固定化生物分子的條件下接觸,並因此提供固定化的生物分子;c)任選地使步驟(b)(i)的固定化的化學修飾的、酶促修飾的或活化的生物分子或步驟(b)(ii)的固定化的生物分子與化學修飾劑、酶促修飾劑或活化劑接觸,以提供固定化的化學修飾的、酶促修飾的和/或活化的生物分子;d)任選地用緩衝液洗滌步驟(b)(i)的固定化的化學修飾的、酶促修飾的或活化的生物分子;步驟(b)(ii)的固定化的生物分子;或步驟(c)的固定化的化學修飾的、酶促修飾的和/或活化的、固定化的生物分子,以去除多餘的或未反應的化學修飾劑、酶促修飾劑或多餘的或未反應的活化劑,e)任選地重複步驟(c)和步驟(d);f)任選地使藥物組分與化學修飾劑、酶促修飾劑或活化劑接觸,以提供化學修飾的、酶促修飾的或活化的藥物組分;g)(i)當進行步驟(f)時,使固定化的生物分子或固定化的化學修飾的、酶促修飾的和/或活化的生物分子與步驟(f)的化學修飾的、酶促修飾的或活化的藥物組分接觸,以形成固定化的生物分子-藥物-綴合物;或(ii)當不進行步驟(f)時,使固定化的生物分子或固定化的化學修飾的、酶促修飾的和/或活化的生物分子與藥物組分接觸,以形成固定化的生物分子-藥物-綴合物;h)任選地用緩衝液洗滌步驟(g)的固定化的生物分子-藥物-綴合物,以去除多餘的或未反應的試劑,以提供純化的固定化的生物分子-藥物綴合物;i)從捕獲樹脂釋放純化的生物分子-藥物-綴合物;其中生物分子為抗體、修飾的抗體或抗體片段。本發明的以上方法的關鍵特徵為在該方法中採用的捕獲樹脂能夠以一致且可再現的方式固定化生物分子。捕獲樹脂對生物分子的一致固定化應該導致通過以上方法產生的所得生物分子-藥物-綴合物的降低的變化。例如,藥物組分在其處被附接至固定化的生物分子的點的變化可能被降低,因此導致藥物組分與固定化的生物分子之間更一致的附接點。區域特異性中的此類改進將為改進所得生物分子-藥物-綴合物產物的一致性方面所期望的。以上方法的期望特徵為生物分子的固定化降低分子間相互作用並因此降低聚集。對於具有三級結構的複雜生物分子諸如抗體,固定化到捕獲樹脂通過捕捉樹脂對生物分子的多點附接使展開最小化。因此,樹脂和生物分子之間的附接點的數目與通過固定化步驟實現的穩定性的增強良好相關。與採用親本蛋白a、蛋白g或蛋白l作為生物分子捕獲部分相比,採用基於非肽的蛋白a、蛋白g或蛋白l模擬物作為生物分子捕獲部分可以由於模擬物相比於常規的基於蛋白a、蛋白g或蛋白l的系統的增加的區域特異性導致生物分子的固定化中的一致性的相對改進。在生物分子對蛋白的固定化的區域特異性低的情況下,採用親本蛋白a、蛋白g或蛋白質l作為生物分子捕獲部分將固有地導致捕獲樹脂對生物分子的可變的固定化。例如,親本蛋白a、蛋白g或蛋白l可以經由蛋白上的其他位點顯示非特異性結合,這可能使總體相互作用複雜化。如以上解釋的,本發明設想的捕獲樹脂對生物分子的一致固定化然後可以導致通過以上方法產生的所得生物分子-藥物-綴合物的降低的變化。這將為有利的。本發明的樹脂系統的另一個優點存在於,與用於常規的基於蛋白a、蛋白g或蛋白質l的系統的情況相比,更寬範圍的藥物原則上能夠與樹脂綴合。例如,在疏水分子的情況下,可發生在基於親本蛋白a、蛋白g或蛋白l的系統中的其他非特異性結合可破壞或防止此類藥物的有效綴合。在一個實施方案中,捕獲樹脂為非蛋白質捕獲樹脂。在一個實施方案中,捕獲樹脂的生物分子捕獲部分具有以下的分子量:約1000da或更小,任選地約500da或更小、約300da或更小或約200da或更小。在一個實施方案中,捕獲樹脂為非蛋白質捕獲樹脂,並且捕獲樹脂的生物分子捕獲部分具有約1000da或更小的分子量。在另外的實施方案中,捕獲樹脂為基於非肽的捕獲樹脂,並且捕獲樹脂的生物分子捕獲部分具有約1000da或更小的分子量。與採用親本蛋白a、蛋白g或蛋白l或基於肽的蛋白a、蛋白g或蛋白l作為生物分子捕獲部分相比,採用基於非肽的蛋白a、蛋白g或蛋白l模擬物的另一個益處為,模擬物生物分子捕獲部分與廣範圍的常見抗體綴合化學(chemistry)相容,並且可以擴大至工業水平。這與基於蛋白a、蛋白g或蛋白l的生物分子捕獲部分和基於肽的蛋白a、蛋白g或蛋白l捕獲部分形成對比。例如,通常期望靶向固定化的抗體上的賴氨醯側鏈官能基團。目前在臨床發展中的28種抗體藥物綴合物中,近一半(下表中的灰色陰影的那些)採用賴氨酸定向的綴合化學。蛋白a、g或l表面上的固定化配體的蛋白質性質將導致無意的靶向蛋白捕獲樹脂上的賴氨醯側鏈官能基團。蛋白a(swiss-protp02976)具有59個賴氨酸殘基,蛋白g(swiss-protp919909)具有59個賴氨酸殘基,且蛋白l(swiss-protq51918)具有132個賴氨酸殘基。除了如以上描述的配體和抗體賴氨醯殘基之間的競爭以外,基於蛋白a、g和l的捕獲樹脂還存在其他問題。這些包括蛋白的浸提和浸提的加合物的免疫原性。這意味著在製備過程結束之前不能採用這些親和支持物(用於純化或綴合)。由採用基於蛋白a、g和l的捕獲樹脂的此類方法提供的任何綴合物質將不符合目前關於抗體純化和產品質量的監管指導。用於合成生物分子-藥物-綴合物的方法根據本發明,提供了用於合成生物分子-藥物-綴合物的方法。本發明的方法在下面的示意圖中被描述。下面的示意圖中的公開內容不限制本發明的權利要求和內容,而為用作可以使用本發明的說明性方法的視覺指示符。合成化學或酶促修飾的或活化的、固定化的生物分子的方法:根據本發明,提供了一種合成化學或酶促修飾的或活化的、固定化的生物分子的方法,該方法包括:(a)任選地使生物分子與化學修飾劑、酶促修飾劑或活化劑接觸,以提供化學修飾的、酶促修飾的或活化的生物分子;(b)(i)當進行步驟(a)時,使步驟(a)的化學修飾的、酶促修飾的或活化的生物分子與包含基於非肽的蛋白a、蛋白g或蛋白l模擬物生物分子捕獲部分的捕獲樹脂在適於固定化化學修飾的、酶促修飾的或活化的生物分子的條件下接觸,並因此提供固定化的化學修飾的、酶促修飾的或活化的生物分子;或(ii)當不進行步驟(a)時,使生物分子與包含基於非肽的蛋白a、蛋白g或蛋白l模擬物生物分子捕獲部分的捕獲樹脂在適於固定化生物分子的條件下接觸,並因此提供固定化的生物分子;(c)使步驟(b)(i)的固定化的化學修飾的、酶促修飾的或活化的生物分子或步驟(b)(ii)的固定化的生物分子與化學修飾劑、酶促修飾劑或活化劑接觸,以提供固定化的化學修飾的、酶促修飾的和/或活化的生物分子;其中生物分子為抗體、修飾的抗體或抗體片段。蛋白且更具體的抗體的綴合經常用於研究、診斷和治療中。bioconjugatetechniques,第二版(gregthermanson)提供關於創建標記的或綴合的分子的化學、試劑系統和實際應用的高度詳細的信息。它還詳細地描述了關於數百種商購可得的試劑的數十種反應和這些試劑用於修飾或交聯肽和蛋白、糖和多糖、核酸和寡核苷酸、脂質和合成聚合物的用途。下面提供了應用於抗體的關鍵綴合化學的概述。在天然鏈間二硫鍵還原之後與游離硫醇的綴合為抗體綴合和商業adc採用的化學的常用方法。方法包括使抗體與還原劑諸如tcep、dtt、巰基乙胺或本領域熟知的其他合適的還原劑接觸,隨後與式d-x的藥物、配體、標記物綴合,其中d為藥物、配體或標記物並且x為選自馬來醯亞胺、滷代烷、吡啶基二硫化物、烯、乙烯基碸、雙碸(bis-sulphones)、丙烯酸酯、甲基丙烯酸酯和本領域已知的其他硫醇反應性化學的反應性基團。與抗體硫醇綴合的替代方法為(遺傳地)工程化抗體中特定位點處的反應性半胱氨酸殘基,以使藥物、配體或標記物以定義的化學計量綴合而不破壞鏈間二硫鍵。工程化的半胱氨酸通常作為半胱氨酸或穀胱甘肽的混合二硫化物存在。通過完全還原隨後滲濾去除加合物。這破壞鏈間二硫鍵,其必須通過用空氣、cuso4或脫氫抗壞血酸氧化來重新形成。另一個常見的綴合位點為存在於賴氨酸殘基側鏈上的氨基基團。最簡單的方法包括使抗體與式d-y的藥物、配體、標記物或接頭接觸。d具有與以上相同的定義,並且y為選自異氰酸酯、nhs酯、磺醯氯化物(sulfonylchlorides)、環氧化物和本領域技術人員已知的其他試劑的反應性基團。還通常採用與賴氨酸的間接綴合。首先用異雙功能接頭活化賴氨酸側鏈的氨基基團,然後將其與包含互補反應性化學的藥物、配體或標記物綴合。此類偶聯體(couplet)的實例包括用2-亞氨基硫烷(2-iminothiolane)修飾賴氨酸,以創建新的硫醇,隨後與以上描述的硫醇反應性藥物接頭(d-x)中任一種綴合。另一種偶聯體為用異雙功能交聯劑smcc修飾賴氨酸,以創建賴氨酸結合的馬來醯亞胺,隨後與包含配體或標記物游離硫醇的藥物綴合。對於可用於間接賴氨酸綴合的潛在偶聯體的完整綜述,參見hermanson和pierce/thermoscientific交聯劑目錄。幾個組已經開發了摻入具有化學正交於蛋白中20種成蛋白(proteogenic)胺基酸側鏈的側鏈的非天然胺基酸的方法。redwoodbioscience(www.redwoodbioscience.com)已經開發了一種他們稱為醛加標籤(aldehydetagging)的技術。在此他們利用稱為甲醯甘氨酸酶(fge)的天然酶,其通常將高度保守的13胺基酸序列中的cys殘基轉化為i型硫酸酯酶中的甲醯甘氨酸(醛)(wu等人,pnas,2009,106,9,3001)。待與此類修飾的抗體綴合的藥物、配體或標記物必須包含醛反應性化學諸如羥基胺或肼。醛反應性官能團的充分公開可以見於hermanson和perbio目錄中。ambryx開發了一種他們稱為eucode的技術(liu等人,anu.rev.biochem.,2010,79,413)。eucode為一種藉以通過在表達系統中包含三種非天然組分將細胞工程化為將非天然胺基酸摻入外源蛋白中的平臺:1.補充到介質中的非天然胺基酸2.正交氨醯-trna合成酶(aars)3.正交trna正交aars/trna對已經被工程化/選擇為促進在琥珀終止密碼子處的閱讀,並在該位置處摻入非天然胺基酸。使用這種方法,已經將多達70個nnaa摻入到蛋白中。下圖展示了正交胺基酸側鏈和反應性化學的可能組合(從2012年2月在hansonwadeadc峰會上的ambryx展示改編)。sutro已經描述了使用開放、無細胞合成(open,cell-freesynthesis)(ocfs)技術產生抗體和細胞因子。ocfs的特徵為能夠將用可以針對特定密碼子的裝載trna(chargedtrna)將非天然胺基酸摻入到蛋白,以將非天然胺基酸遞送至蛋白上的特定位置—使蛋白適於特定修飾或賦予新的期望的性能(goerke等人,biotechnol.bioeng.,2008,99:351-367)。固定化抗體綴合與所有非天然胺基酸側鏈和互補反應性化學相容,但有一個條件。抗體捕獲配體不得包含作為非天然胺基酸側鏈的部分摻入的新穎化學。用高碘酸鈉氧化糖蛋白中的多糖殘基提供了產生反應性醛基團以用於隨後與包含胺或醯肼的分子、藥物、配體或標記物綴合的溫和且高效的方法。該方法涉及首先使抗體與高碘酸鈉接觸,並且然後與選自胺、醯肼、氨氧基(aminoxy)或其他本領域已知的醛反應性化學的反應性基團綴合。綴合步驟通常在酸性條件下進行以形成肟和腙鍵。在相關方法中,肼基-異-pictet-spengler(hydrazino-iso-pictet-spengler)(hips)連接還將反應性醛基團與取代的肼綴合以形成穩定的氮雜咔啉(azacarboline)綴合物。步驟(a):在一個實施方案中,進行步驟(a)。在一個替代實施方案中,省略步驟(a)。在實施方案中,使生物分子與化學修飾劑、酶促修飾劑或活化劑接觸以提供修飾的或活化的生物分子的步驟涉及還原生物分子。在一個實施方案中,生物分子的還原涉及完全還原。在一個實施方案中,生物分子的還原涉及部分還原。在一個實施方案中,生物分子的還原涉及完全還原,隨後再氧化。在一個實施方案中,通過使生物分子與還原劑諸如三(2-羧乙基)膦(tcep)、二硫蘇糖醇(dtt)、巰基乙胺或其他合適的還原劑接觸來還原生物分子。優選地還原劑為三(2-羧乙基)膦(tcep)。在一個實施方案中,還原的生物分子通過使其與氧化劑諸如空氣、cuso4或脫氫抗壞血酸(dhaa)接觸來再氧化。優選地氧化劑為脫氫抗壞血酸(dhaa)。在一個實施方案中,還原生物分子的過程在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。在一個實施方案中,還原生物分子的過程在以下的ph下進行:約5至約10、優選地約7至約8、優選地約7.4。在一個實施方案中,還原生物分子的過程在螯合劑諸如edta的存在下進行。在一個實施方案中,還原生物分子的過程涉及將生物分子與還原劑孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天,並且還任選地約6小時至約18小時。在一個實施方案中,使生物分子與化學修飾劑、酶促修飾劑或活化劑接觸以提供修飾的或活化的生物分子的步驟涉及使生物分子與交聯劑部分(crosslinkermoiety)反應。例如,交聯劑部分可以為胺與巰基交聯劑(amine-to-sulfhydrylcrosslinker),例如在相對的末端具有nhs-酯和馬來醯亞胺反應性基團的交聯劑。這種修飾或活化生物分子的方法有效地產生生物分子-接頭-藥物-綴合物。合適的交聯劑通常能夠與藥物基團上的伯胺基團反應(經由反應性nhs酯末端),並且還與生物分子上的半胱氨酸殘基反應(經由反應性馬來醯亞胺末端)。在該特定實例中,馬來醯亞胺末端將與固定化的生物分子中的半胱氨酸反應。此類交聯劑的實例為4-(n-馬來醯亞胺基甲基)環己烷-1-羧酸琥珀醯亞胺酯(succinimidyl4-(n-maleimidomethyl)cyclohexane-1-carboxylate)(smcc)。在一個實施方案中,與交聯劑反應的過程在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。可選擇地,與交聯劑反應的過程在包含磷酸鈉緩衝液、nacl和螯合劑諸如edta的『修飾緩衝液』中進行。在一個實施方案中,與交聯劑反應的過程在以下的ph下進行:約7至約9、優選地約7至約8、優選地約8.0。在一個實施方案中,與交聯劑反應的過程在螯合劑諸如edta的存在下進行。在一個實施方案中,與交聯劑反應的過程涉及將生物分子與交聯劑孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天,並且還任選地約6小時至約18小時。在一個實施方案中,使生物分子與化學修飾劑或活化劑接觸以提供修飾的或活化的生物分子的步驟涉及使生物分子與特勞特試劑(traut’sreagent)反應。在一個實施方案中,與特勞特試劑反應的過程在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。在一個實施方案中,與特勞特試劑反應的過程在以下的ph下進行:約7至約9、優選地約7至約8、優選地約8.0。在一個實施方案中,與特勞特試劑反應的過程在螯合劑諸如edta的存在下進行。在一個實施方案中,與特勞特試劑反應的過程涉及將生物分子與還原劑孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天,並且還任選地約6小時至約18小時。在一個實施方案中,將活化的生物分子洗滌以去除任何修飾/活化劑。在一個實施方案中,洗滌涉及用緩衝液潤洗,任選地其中緩衝液為磷酸鹽緩衝鹽水(pbs)。其他合適的緩衝液包括:磷酸鉀緩衝液;磷酸鈉緩衝液;檸檬酸鈉緩衝液;bis-tris丙烷緩衝液;hepes緩衝液;乙酸鈉緩衝液;檸檬酸鈉緩衝液;二甲胂酸緩衝液;乙酸銨緩衝液;咪唑緩衝液;n-二羥乙基甘氨酸緩衝液;和2-(n-嗎啉代)乙磺酸(mes)緩衝液。例如,可以將生物分子用在以下的ph下的緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)進行洗滌:約7至約8,優選地約7.4。任選地,活化的生物分子的潤洗在螯合劑諸如edta的存在下進行。潤洗活化的生物分子的另一個實例涉及用緩衝液諸如pbs,隨後是包括檸檬酸鈉、nacl和螯合劑諸如edta的『綴合緩衝液』潤洗樹脂。步驟(b):當進行步驟(a)時,步驟(b)涉及使步驟(a)的化學修飾的、酶促修飾的或活化的生物分子與包含基於非肽的蛋白a、蛋白g或蛋白l模擬物生物分子捕獲部分的捕獲樹脂在適於固定化化學修飾的、酶促修飾的或活化的生物分子的條件下接觸,並因此提供固定化的化學修飾的、酶促修飾的或活化的生物分子。當省略步驟(a)時,步驟(b)涉及使生物分子與包含基於非肽的蛋白a、蛋白g或蛋白l模擬物生物分子捕獲部分的捕獲樹脂在適於固定化生物分子的條件下接觸,並因此提供固定化的生物分子。在一個實施方案中,使生物分子與捕獲樹脂接觸的步驟包括將生物分子與捕獲樹脂孵育。孵育可以在以下進行:在約0℃至約100℃的溫度下,優選地在約5℃至約50℃的溫度下且任選地在約10℃至約40℃的溫度下。理想地,孵育在約15℃至約37℃的溫度下進行,例如孵育在室溫,諸如約21℃下進行。可選擇地,孵育在約37℃下進行。孵育可以進行約1分鐘至約3天的時間段,例如約10分鐘至約18小時的時間段。優選地孵育進行約20分鐘至約1小時的時間段。在一個實施方案中,孵育在水性介質中進行。在一個替代實施方案中,孵育在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)或與期望的結合ph和化學相容的任何緩衝鹽中進行,任選地孵育在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。在一個實施方案中,使用包括溶劑諸如dmso、dma或dmf的共溶劑進行孵育。共溶劑可以在0.5-80%v/v,諸如0.5-50%v/v的範圍內存在。在一個實施方案中,孵育在以下的ph下進行:約5至約10、優選地約5至約8、更優選地約6至約8。在優選的實施方案中,孵育在約6至約7.5的ph下,理想地在約6.5的ph下進行。在另一個優選的實施方案中,孵育在約7至約8的ph下,理想地在約7.4的ph下進行。這導致改進的抗體對衍生化支持物結合。在一個實施方案中,洗滌固定化的生物分子(即被固定化在捕獲樹脂上的生物分子)以去除未固定化在捕獲樹脂上的任何生物分子。固定化的生物分子的洗滌可以通過用新鮮溶劑潤洗實現。例如,固定化的生物分子的洗滌可以通過用緩衝溶液諸如pbs潤洗實現。任選地,固定化的生物分子的潤洗在螯合劑諸如edta的存在下進行。可選擇地,固定化的生物分子的洗滌可以通過用包括磷酸鈉緩衝液、nacl和螯合劑諸如edta的『修飾緩衝液』潤洗實現。步驟(c):在一個實施方案中,進行步驟(c)。在一個替代實施方案中,省略步驟(c)。步驟(c)涉及使步驟(b)(i)的固定化的化學修飾的、酶促修飾的或活化的生物分子或步驟(b)(ii)的固定化的生物分子與化學修飾劑、酶促修飾劑或活化劑接觸,以提供固定化的化學修飾的、酶促修飾的和/或活化的生物分子。在一個實施方案中,使固定化的生物分子與化學修飾劑或活化劑接觸以提供修飾的或活化的、固定化的生物分子的步驟涉及還原生物分子。在一個實施方案中,生物分子的還原涉及完全還原。在一個實施方案中,生物分子的還原涉及部分還原。在一個實施方案中,生物分子的還原涉及完全還原,隨後再氧化。在一個實施方案中,通過使生物分子與還原劑諸如三(2-羧乙基)膦(tcep)、二硫蘇糖醇(dtt)、巰基乙胺或其他合適的還原劑接觸來還原生物分子。優選地還原劑為三(2-羧乙基)膦(tcep)。在一個實施方案中,還原的生物分子通過使其與氧化劑諸如空氣、cuso4或脫氫抗壞血酸(dhaa)接觸來再氧化。優選地氧化劑為脫氫抗壞血酸(dhaa)。在一個實施方案中,還原生物分子的過程在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。在一個實施方案中,還原生物分子的過程在以下的ph下進行:約5至約10、優選地約7至約8、優選地約7.4。在一個實施方案中,還原生物分子的過程在螯合劑諸如edta的存在下進行。在一個實施方案中,還原生物分子的過程涉及將生物分子與還原劑孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天,並且還任選地約6小時至約18小時。在一個實施方案中,使固定化的生物分子與化學修飾劑、酶促修飾劑或活化劑接觸以提供修飾的或活化的、固定化的生物分子的步驟涉及使生物分子與交聯劑部分反應。例如,交聯劑部分可以為胺與巰基交聯劑,例如在相對末的端具有nhs-酯和馬來醯亞胺反應性基團的交聯劑。這種修飾或活化生物分子的方法有效地產生生物分子-接頭-藥物-綴合物。合適的交聯劑通常能夠與藥物上的伯胺基團反應(經由反應性nhs酯末端),並且還與生物分子上的半胱氨酸殘基反應(經由反應性馬來醯亞胺末端)。在該特定實例中,馬來醯亞胺末端將與固定化的生物分子中的半胱氨酸反應。此類交聯劑的實例為4-(n-馬來醯亞胺基甲基)環己烷-1-羧酸琥珀醯亞胺酯(smcc)。在一個實施方案中,與交聯劑反應的過程在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。可選擇地,與交聯劑反應的過程在包括磷酸鈉緩衝液、nacl和螯合劑諸如edta的『修飾緩衝液』中進行。在一個實施方案中,與交聯劑反應的過程在以下的ph下進行:約7至約9、優選地約7至約8、優選地約8.0。在一個實施方案中,與交聯劑反應的過程在螯合劑諸如edta的存在下進行。在一個實施方案中,與交聯劑反應的過程涉及將生物分子與交聯劑孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天,並且還任選地約6小時至約18小時。在一個實施方案中,使固定化的生物分子與化學修飾劑或活化劑接觸以提供修飾的或活化的、固定化的生物分子的步驟涉及使生物分子與特勞特試劑反應。在一個實施方案中,與特勞特試劑反應的過程在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。在一個實施方案中,與特勞特試劑反應的過程在以下的ph下進行:約7至約9、優選地約7至約8、優選地約8.0。在一個實施方案中,與特勞特試劑反應的過程在螯合劑諸如edta的存在下進行。在一個實施方案中,與特勞特試劑反應的過程涉及將生物分子與還原劑孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天,並且還任選地約6小時至約18小時。步驟(d):在一個實施方案中,進行步驟(d)。在一個替代實施方案中,省略步驟(d)。在一個實施方案中,洗滌步驟(b)(i)的固定化的化學修飾的、酶促修飾的或活化的生物分子;步驟(b)(ii)的固定化的生物分子;或步驟(c)的固定化的化學修飾的、酶促修飾的和/或活化的、固定化的生物分子,以去除任何修飾劑/活化劑。在一個實施方案中,洗滌涉及用緩衝液潤洗,任選地其中緩衝液為磷酸鹽緩衝鹽水(pbs)。其他合適的緩衝液包括:磷酸鉀緩衝液;磷酸鈉緩衝液;檸檬酸鈉緩衝液;bis-tris丙烷緩衝液;hepes緩衝液;乙酸鈉緩衝液;檸檬酸鈉緩衝液;二甲胂酸緩衝液;乙酸銨緩衝液;咪唑緩衝液;n-二羥乙基甘氨酸緩衝液;和2-(n-嗎啉代)乙磺酸(mes)緩衝液。例如,可以將固定化的生物分子用在以下的ph下的緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)進行洗滌:約7至約8,優選地約7.4。任選地,活化的、固定化的生物分子的潤洗在螯合劑諸如edta的存在下進行。潤洗活化的、固定化的生物分子的另一個實例涉及用緩衝液諸如pbs,隨後是包括檸檬酸鈉、nacl和螯合劑例如edta的『綴合緩衝液』潤洗樹脂。步驟(e):在一個實施方案中,重複步驟(c)一次、兩次或三次。在一個實施方案中,重複步驟(c)一次。在一個實施方案中,重複步驟(c)兩次。在一個實施方案中,重複步驟(c)三次。在一個實施方案中,重複步驟(d)一次、兩次或三次。在一個實施方案中,重複步驟(d)一次。在一個實施方案中,重複步驟(d)兩次。在一個實施方案中,重複步驟(d)三次。步驟(f):在一個實施方案中,進行步驟(f)。在一個替代實施方案中,省略步驟(f)。步驟(f)涉及使藥物組分與化學修飾劑、酶促修飾劑或活化劑接觸,以提供化學修飾的、酶促修飾的和/或活化的藥物組分。在一個實施方案中,使藥物組分與化學修飾劑、酶促修飾劑或活化劑接觸以提供修飾的或活化的藥物組分的步驟涉及使藥物組分與交聯劑部分反應。例如,交聯劑部分可以為胺與巰基交聯劑,例如在相對的末端具有nhs-酯和馬來醯亞胺反應性基團的交聯劑。這種修飾或活化藥物組分的方法有效地產生生物分子-接頭-藥物-綴合物。合適的交聯劑通常能夠與生物分子上的半胱氨酸殘基反應,例如化學修飾的、酶促修飾的或活化的生物分子(經由反應性馬來醯亞胺末端),並且還與藥物組分上的胺部分反應(經由反應性nhs酯末端)。在該特定實例中,馬來醯亞胺末端將與固定化的生物分子中的半胱氨酸反應。此類交聯劑的實例為4-(n-馬來醯亞胺基甲基)環己烷-1-羧酸琥珀醯亞胺酯(smcc)。在一個實施方案中,與交聯劑反應的過程在緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)中進行。可選擇地,與交聯劑反應的過程在包括磷酸鈉緩衝液、nacl和螯合劑例如edta的『修飾緩衝液』中進行。在一個實施方案中,與交聯劑反應的過程在以下的ph下進行:約7至約9、優選地約7至約8、優選地約8.0。在一個實施方案中,與交聯劑反應的過程在螯合劑諸如edta的存在下進行。在一個實施方案中,與交聯劑反應的過程涉及將藥物組分與交聯劑孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天,並且還任選地約6小時至約18小時。步驟(g):步驟(g)涉及使固定化的生物分子或固定化的化學修飾的、酶促修飾的和/或活化的生物分子與步驟(f)的化學修飾的、酶促修飾的或活化的藥物組分接觸(當進行步驟(f)時)或使固定化的生物分子或固定化的化學修飾的、酶促修飾的和/或活化的生物分子與藥物組分接觸以形成固定化的生物分子-藥物-綴合物。在一個實施方案中,使固定化的生物分子或化學修飾的、酶促修飾的和/或活化的、固定化的生物分子與步驟(f)的化學修飾的、酶促修飾的或活化的藥物組分接觸的步驟(當進行步驟(f)時)涉及同時(1)進行藥物組分的化學修飾、酶修飾或活化和(2)與固定化的生物分子或化學修飾的、酶促修飾的和/或活化的、固定化的生物分子接觸。換言之,使生物分子在其被原位產生時與化學修飾的、酶促修飾的或活化的藥物組分接觸。在該實施方案中,步驟(f)和(g)不為單獨的步驟,而為單個的、組合的步驟。在一個實施方案中,使固定化的生物分子或化學修飾的、酶促修飾的和/或活化的、固定化的生物分子與藥物組分接觸以形成固定化的生物分子-藥物-綴合物的步驟涉及使化學修飾的、酶促修飾的和/或活化的、固定化的生物分子與藥物組分在緩衝溶液中接觸,如上文關於步驟(c)描述的。在一個實施方案中,使固定化的生物分子或化學修飾的、酶促修飾的或活化的、固定化的生物分子與藥物組分接觸以形成固定化的生物分子-藥物-綴合物的步驟涉及在以下ph下使化學修飾的、酶促修飾的或活化的、固定化的生物分子與藥物組分接觸:約5至約8、優選地約7至約8且更優選地約7.4。在一個實施方案中,使固定化的生物分子或化學修飾的、酶促修飾的或活化的、固定化的生物分子與藥物組分接觸以形成固定化的生物分子-藥物-綴合物的步驟在螯合劑諸如edta的存在下進行。在一個實施方案中,使固定化的生物分子或化學修飾的、酶促修飾的或活化的、固定化的生物分子與藥物組分接觸以形成固定化的生物分子-藥物-綴合物的步驟涉及使化學修飾的、酶促修飾的或活化的、固定化的生物分子與藥物組分孵育以下的時間段:約20分鐘至約3天,任選地,約1小時至約2天並且還任選地約6小時至約18小時。步驟(h):在一個實施方案中,進行步驟(h)。在一個替代實施方案中,省略步驟(h)。在一個實施方案中,在從捕獲樹脂釋放生物分子-藥物-綴合物的步驟之前,洗滌固定化的生物分子-藥物-綴合物。該洗滌去除任何未反應的藥物組分。在一個實施方案中,洗滌涉及用緩衝液潤洗,任選地其中緩衝液為磷酸鹽緩衝鹽水(pbs)和其他溶劑。其他合適的緩衝液包括:磷酸鉀緩衝液;磷酸鈉緩衝液;檸檬酸鈉緩衝液;bis-tris丙烷緩衝液;hepes緩衝液;乙酸鈉緩衝液;檸檬酸鈉緩衝液;二甲胂酸緩衝液;乙酸銨緩衝液;咪唑緩衝液;n-二羥乙基甘氨酸緩衝液;和2-(n-嗎啉代)乙磺酸(mes)緩衝液。例如,可以將固定化的生物分子-藥物-綴合物用在以下的ph下的緩衝溶液諸如磷酸鹽緩衝鹽水(pbs)和二甲基乙醯胺(dma)進行洗滌:約5至約7。任選地,固定化的生物分子-藥物-綴合物的潤洗在螯合劑諸如edta的存在下進行。在一個實施方案中,在用緩衝液從捕獲樹脂釋放純化的綴合物的步驟之前,洗滌固定化的綴合物,任選地其中緩衝液為磷酸鹽緩衝鹽水(pbs)或適用於製劑的其他緩衝液。洗滌去除任何剩餘的或多餘的有機溶劑諸如dmso、dma或dmf。步驟(i):在一個實施方案中,從捕獲樹脂釋放生物分子-藥物-綴合物的步驟涉及:a)將支持物-生物分子化合物暴露於釋放劑(releaseagent);和/或b)改變ph以破壞支持物-生物分子鍵。在一個實施方案中,釋放劑為氫鍵破壞物(hydrogenbonddisrupter),諸如六氟異丙醇(hexafluoroisopropanol)、2,2,2-三氟乙醇(trifluoroethanol)或二甲基亞碸(dmso)的共溶劑。在一個實施方案中,將釋放劑與支持物-生物分子孵育。孵育可以在以下進行:在約0℃至約100℃的溫度下,優選地在約5℃至約50℃的溫度下且任選地在約10℃至約40℃的溫度下。理想地,孵育在約15℃至約37℃的溫度下進行,例如孵育在室溫,諸如約21℃下進行。可選擇地,孵育在約37℃下進行。孵育可以進行約1分鐘至約3天的時間段。優選地孵育進行約30分鐘至約2小時的時間段。孵育可以在水性介質中進行。可選擇地,孵育可以在溶劑諸如dmf、dma、dmso、meoh或mecn中進行。可選擇地,孵育可以在具有多達80%溶劑、優選地0.5%至50%且最優選0.5%至10%v/v的水性-溶劑混合物中進行。在某些情況下,一種或更多種以上溶劑(包括水)的混合物可以為合適的。必要時還可以包括穩定劑以確保綴合物保持完整。在一個實施方案中,從捕獲樹脂釋放生物分子-藥物-綴合物的步驟涉及改變ph。可以通過任何足以破壞支持物-生物分子鍵但將不影響生物分子的活性、完整性或3d結構的量改變ph。例如,可以調節ph使其為酸性。在一個實施方案中,將ph從約ph6降低至約ph2。任選地,將ph調節至小於約ph5,例如約ph3至約5,例如小於約ph4。在一個實施方案中,將ph降低至約ph3。可選擇地,可以調節ph使其為鹼性。在一個實施方案中,將ph增加至約ph8至約ph10。任選地,將ph調節至大於ph8。例如,可以將ph增加至約ph9。可以將ph增加至大於ph9。例如,可以將ph增加至約ph10。可以將ph增加至大於ph10,但通常會低於ph14。生物分子-藥物-綴合物可以經歷用釋放劑一次或更多次處理。有利地,用新鮮釋放劑的第二次或隨後處理的使用可以導致從捕獲樹脂釋放的生物分子-藥物-綴合物的量增加。新鮮釋放劑為先前未與固定化的生物分子-藥物-綴合物孵育過的釋放劑。在一個實施方案中,從捕獲樹脂釋放生物分子-藥物-綴合物的步驟涉及使生物分子-藥物-綴合物與鹽接觸。例如,可以使生物分子-藥物-綴合物與nacl接觸。鹽的濃度可以為約0.1m至約10m,優選地約0.1m至約1m。在一個實施方案中,洗脫的生物分子-藥物-綴合物在從捕獲樹脂釋放綴合物的步驟之後被中和。例如,綴合物可以被捕獲到2%v/v的1m三(羥甲基)氨基乙烷(tris)中。洗滌步驟:在一個實施方案中,在本發明的方法中洗滌中間體的步驟包括去除不與捕獲樹脂結合的物質,諸如汙染物。典型的汙染物包括用於活化固定化的生物分子的過量試劑、未被固定化在捕獲樹脂上的生物分子和未與活化的、固定化的生物分子反應的藥物組分或多餘的剩餘的溶劑或共溶劑。不影響生物分子的活性、完整性或3d結構或固定化的生物分子與捕獲樹脂之間的結合的完整性的任何介質都可以用於洗滌中間體。優選地,緩衝液為等滲的並且通過模擬生理ph和離子強度來誘導對生物分子諸如抗體的穩定環境。在一個實施方案中,通過過濾洗滌活化的、固定化的生物分子。任選地,將所得濾液例如通過使用膜濾芯(membranecartridge)離心進行緩衝液交換。通常,將添加劑引入緩衝介質。這些添加劑對緩衝系統和包含於其中的生物分子誘導了一定水平的控制。例如,添加劑諸如tris或組氨酸被引入至緩衝的工藝流中以維持ph並最小化偶然的酸化。通常,生物分子工藝流的ph應該被維持在ph5和9.5之間,並且避免長時間處於ph限值的極端值。可以添加無機鹽諸如0.1mnacl(aq)以維持工藝流的離子強度。離子和非離子去垢劑諸如吐溫(聚山梨醇酯)可以被添加至緩衝液中以有利地增加難溶性生物分子在緩衝介質中的溶解度並最小化聚集。包含捕獲樹脂和活化劑的混合物:根據本發明,提供了一種混合物,所述混合物包含:(i)捕獲樹脂,所述捕獲樹脂包含選自由基於非肽的蛋白a、蛋白g或蛋白l模擬物組成的組的抗體、修飾的抗體或抗體片段捕獲部分;和(ii)化學修飾劑或活化劑。在一個實施方案中,捕獲樹脂在其表面上包括固定化的抗體、修飾的抗體或抗體片段。捕獲樹脂在合成生物分子-藥物-綴合物中的用途:根據本發明,提供了包含選自由基於非肽的蛋白a、蛋白g或蛋白l模擬物組成的組的抗體、修飾的抗體或抗體片段捕獲部分的捕獲樹脂在合成生物分子-藥物-綴合物中的用途。捕獲樹脂:多年來,研究人員已經嘗試開發對一系列全長抗體、片段或融合物具有親和性的配體,作為傳統的蛋白a、g或l親和純化支持物的替代物。成功的配體發現/開發的主要標準為:1.提供高的初始純化的高選擇性抗體2.有用的動態結合能力3.與保留抗體完整性相容的洗脫條件4.支持物在多個洗脫/清洗循環期間的穩定性5.相對於蛋白a、g或l支持物的降低的成本在使用這些配體用於固相抗體綴合的場景下,以上標準1不是關鍵的,因為綴合過程開始於純化的抗體。然而,配體必須完全符合剩餘的4個標準。另外,配體理想地必須具有與抗體相互作用的確定的位點,該確定的位點提供合適的親和結合強度以用於綴合。該屬性為必要的,使得抗體可以在緩衝液補充期間隨時間結合至支持物而不無意地被洗脫。另外,相互作用的確定位點為期望的,以推斷結合的抗體複合物向周圍溶液相的一致的構象展示以及提供用於一致和可再現的綴合化學的工具的作用。抗體為具有許多用於親和純化的充分確定的結合結構域的充分表徵的生物分子。第一個定義的區域為蛋白a和蛋白g結合口袋,其用於使用蛋白a/g和蛋白a/g模擬物支持物的親和層析中。蛋白a與fc區域中的ch2ch3鏈間結構域經由與以下胺基酸殘基的一些非共價相互作用而相互作用:thr250、leu251、met252、ile253、his310、gln311、leu314、asn315、lys338、glu345、ala431、leu432、his433、asn434和his435。蛋白a模擬物支持物已經被合理設計為與該結構域經由以上定義的一個或更多個胺基酸相互作用。這些模擬物支持物為igg結合和綴合提供合適的親和配體。蛋白a模擬物支持物可以在亞類中被定義為摻入基於非肽、肽或胺基酸的配體。類似地,蛋白g與fc區域中的ch2ch3鏈間結構域經由與胺基酸殘基ile253、ser254、gln311、glu380、glu382、his433、asn434和his435的一些非共價相互作用而相互作用。蛋白g模擬物支持物已經被合理設計為與該結構域經由以上描述的一個或更多個胺基酸相互作用。再次,這些模擬物支持物為igg結合和綴合提供合適的親和配體。蛋白g模擬物支持物可以在亞類中被定義為摻入基於非肽、肽或胺基酸的配體。在一個實施方案中,捕獲樹脂能夠結合生物分子上的蛋白a或蛋白g結合口袋。蛋白a模擬物的商業實施方案為mabsorbenttma1p、a2p和a3p(prometicbiosciences)。這些親和支持物符合蛋白a模擬物的標準,因為這些非肽支持物模擬蛋白a的疏水核心結構中的phe-132、tyr-133二肽結合位點。第二個定義的區域為被蛋白l親和基質靶向的抗體輕鏈。蛋白l特異性結合κi、ii和iv輕鏈,但不結合κiii和γ輕鏈。已經繪製了蛋白l與抗體之間的相互作用,並且注意到氫鍵和鹽橋在結合中為重要的。蛋白l結構域的總計11個親水胺基酸殘基—即:ala、asp、gln、glu、gly、ile、leu、lys、phe、thr、tyr—在形成這些鍵中為重要的。已經通過使用結構上與以上公開的11個胺基酸(wo2004/035199a)相似的化學化合物創建三嗪支架組合文庫來開發蛋白l模擬物親和支持物。在wo2004/035199a中公開了,蛋白l模擬物被定義為具有蛋白l對抗體或片段的50%的親和性和如通過結合fab片段證明的對輕鏈的特異性的配體。本文公開的或本領域技術人員已知的任何合適的支架可以被替換為三嗪支架,只要對輕鏈的親和性和特異性的特徵被保留。此類樹脂可用於固定化包含κi、ii和iv輕鏈的抗體和片段。蛋白l模擬物的一個商業實施方案為fabsorbenttmf1phf(prometicbiosciences)。該親和支持物符合蛋白l模擬物的標準,但也結合包含γ輕鏈的抗體和片段。因此,該親和支持物廣泛適用於抗體親和結合和綴合。在一個實施方案中,捕獲樹脂能夠結合被蛋白l親和基質靶向的抗體輕鏈。第三個定義的區域為跨越寬範圍物種的所有抗體同種型的fab臂中的保守核苷酸結構域。結合位點包含4個胺基酸殘基,其中第一個為tyr或phe,剩餘的三個為tyr、tyr和trp。儘管結合口袋位置和胺基酸側鏈取向在晶體結構覆蓋物中為保守的,但在從抗體至抗體的總體骨架序列變化中與在編號方案中存在輕微差異。這通過比較商業抗體赫賽汀(herceptin)和利妥昔單抗的保守核苷酸結合位點在下面被證明。已經被合理設計為與該結構域經由以上描述的一個或更多個胺基酸相互作用的核苷酸模擬物(非肽、肽、核苷酸類似物和胺基酸)為用於igg結合和綴合的合適的親和配體。在一個實施方案中,捕獲樹脂能夠結合抗體的fab臂中的保守核苷酸結構域。第四個定義的區域為存在於完整抗體的fc區域的ch2結構域中的asn297上的聚糖結構。作為親和配體的m-氨基苯基硼酸結合糖蛋白分子的糖部分上存在的糖殘基諸如甘露糖、半乳糖或葡萄糖上的順式二醇基團。這種相互作用提供可逆的五元環複合物。典型的抗體聚糖結構在下面示出,突出抗體聚糖中甘露糖和半乳糖的存在(從arnold等人,advancesinexperimentalmedicineandbiology,2005,546,27-43改編)。在一個實施方案中,捕獲樹脂能夠結合存在於完整抗體的fc區域的ch2結構域中的asn297上的聚糖結構。圖2聚糖結構和同種型。a)用於聚糖的一般術語b)igg的主要聚糖結構。g=半乳糖單元,f=巖藻糖單元。在arnold等人之後修改。配體可以被附接至親和層析領域中熟知的一系列固體支持物基質。這些包括例如合成聚合物(諸如聚丙烯醯胺、聚乙烯醇或聚苯乙烯),特別為交聯合成聚合物,無機支持物(諸如基於二氧化矽的支持物)並且特別為多糖支持物(諸如澱粉、纖維素和瓊脂糖)。適用於抗體結合的特異性配體-支持物描述如下:『非肽』蛋白a、g和l模擬物親和支持物結合合成化學文庫篩選的蛋白a、g或l相互作用的分子建模已經使這些蛋白的小分子模擬物的半理性設計成為可能(li等人,naturebiotechnology,1998,16,190-195)。此類樹脂的實例包括商購可得的支持物mabsorbenta1p和fabsorbentf1phf(prometicbiosciences)。mabsorbenta1p、mabsorbenta2phf和fabsorbentf1phf支持物在合成的芳族三嗪支架上形成(www.prometicbioscience.com)。us20010045384公開了基於亞氨基二乙酸酯(iminodiacetate)(ida)型支架組裝的蛋白a模擬物配體-複合物。ida支架用三嗪基(triazyl)配體衍生化以提供多價三嗪基配體-複合物。wo9808603描述了使用親和樹脂從細胞培養物上清液、血清、血漿或初乳中分離免疫球蛋白。這些親和樹脂包含合成的單環或雙環芳族或雜芳族配體,以促進免疫球蛋白純化。作為抗體親和樹脂有希望的另一種配體為磺胺二甲嘧啶。與磺胺二甲嘧啶偶聯的葡聚糖微粒特異地結合抗體(yi等人,prep.biochem.biotechnol.,2012,42,6,598-610)。在選擇以上描述的前導候選配體時,基於缺乏抗體特異性排除許多配體。本文公開了,對於固相抗體綴合樹脂,特異性沒有結合效率、能力和穩定性那麼重要,並且因此這些未被低估。『肽』蛋白a、g或l模擬物親和支持物已經公開了許多蛋白a模擬物肽。menegatti鑑定了具有結合抗體fc區域的序列hwrgwv的六肽(hexapeptide)(menegatti等人,journalofseparationscience,2012,35,22,3139–3148)。fassina等人通過合成和篩選包含具有四齒賴氨酸核心(tetradentatelysinecore)的隨機合成分子組成的合成多聚體肽文庫,鑑定了蛋白a模擬物肽tg191318(fassina等人,j.mol.recognit.,1996,9,564)。ep1997826公開了包含x1-arg-thr-tyr的肽。lund等人公開了適用於抗體親和層析的兩種肽配體(lund等人,jchromatogr.a,2012,1225,158-167)。daag和d2aag包含l-精氨酸、l-甘氨酸和合成芳香酸2,6-二叔丁基-4-羥基苄基丙烯酸酯(dbhba)。胺基酸蛋白a、g或l模擬親和載體除了以上描述的複合大分子配體以外,已經提出簡單胺基酸作為以相同方式結合抗體的蛋白a模擬物(naik等人,j.chromatogr.a,2011,1218,1756-1766)。其實例為absep,一種包含色氨酸的聚甲基丙烯酸酯樹脂,具有對抗體fc區域中的蛋白a結合位點的高親和性。包含胺基酸酪氨酸、組氨酸和苯丙氨酸的樹脂也適用於抗體固定化和綴合(bueno等人,j.chromatogr.b,biomed.appl.,1995;667,1,57-67)。核苷酸結合位點親和支持物開發抗體純化配體的另一個策略已經利用抗體的fab可變區域中較不知名的保守核苷酸結合位點(nbs)(alves等人,anal.chem.,2012,84,7721-7728)。已經將核苷酸類似物吲哚丁酸與toyopearlaf-650-氨基m樹脂偶聯以製備符合以上標準1-5的支持物。可用於抗體親和層析的廣泛範圍的其他核苷酸類似物被描述於wo/2012/099949中。碳水化合物結合樹脂固定化在多種支持物上的配體m-氨基苯基硼酸已被用於純化糖蛋白。配體結合存在於糖蛋白分子(包括抗體)的糖部分內的糖殘基(諸如甘露糖、半乳糖或葡萄糖)上的順式二醇基團,形成可逆的五元環複合物。可以通過降低ph或通過使用包含tris或山梨糖醇的洗脫緩衝液來解離該複合物。捕獲樹脂的配體能夠與生物分子通過配體和生物分子(例如蛋白、抗體、修飾的抗體或抗體片段)之間的特異的、可逆的和非共價鍵的相互作用而相互作用。非共價相互作用可以分類為離子型、範德華氏型、氫鍵型或疏水型。這些相互作用以三維方式起作用以幫助靶生物分子對捕獲樹脂的配體的柔性和構象。當緊密接近配體時,生物分子可以推斷這些相互作用中的一種、幾種或全部,以提供配體-生物分子複合物。配體與生物分子之間的距離和配體的極性和電負性將確定這些相互作用的強度。此外,這些相互作用的強度可以被定義為親和力。配體和生物分子之間的高親和力構成具有增強的穩定性的配體-生物分子複合物(us2009/0240033)。在一個實施方案中,捕獲樹脂包含基於非肽的蛋白a、蛋白g或蛋白l模擬物。捕獲樹脂能夠結合抗體、修飾的抗體或抗體片段。基於非肽的蛋白a、蛋白g或蛋白l模擬物已被用於染料配體層析,其為利用固定化在固體支持物(諸如瓊脂糖)上的共價鍵紡織品染料來純化蛋白的親和層析模式。這些染料類似於蛋白對其具有親和性的天然底物/蛋白配體。這種純化和分離的模式通常被稱為假親和層析(pseudo-affinitychromatography)。染料配體親和層析為非特異性的,但該技術對於多種蛋白的廣結合範圍為有利的。純化技術的進步採用修飾的染料作為蛋白正常底物/配體的競爭性抑制物(p.dan等人,j.chromatography,1979,165,3,301-319)。通常採用基於三嗪基的配體諸如辛巴藍(cibacronblue)3ga、普施安紅(procionred)h-3b、普施安藍mx3g、普施安黃h-a等,並且解決了原始商業染料諸如藍葡聚糖的純度、滲漏和毒性的問題(lowe等人,trendsbiotechnology,1992,10,442-448)。已經成功地將三嗪基配體用於純化白蛋白、氧化還原酶、脫羧酶、糖酵解酶、核酸酶、水解酶、裂解酶、合成酶和轉移酶(n.labrou,methodsmol.biol.2002,147,129-139)。仿生染料配體親和層析的限制為從生物分子至生物分子的親和強度為明顯可變的,並且在許多情況下,為一種蛋白提供強親和強度(affinitystrength)的配體可能不適用於另一種蛋白。因此,通常需要進行廣泛的和憑經驗的篩選過程以鑑定對感興趣的生物分子具有期望親和性的合適的合成配體。因此,為了幫助結構化地闡明影響對生物分子的親和結合的合適的配體,已結合配體自支持物的剛性空間分離將多價支架基序摻入到配體結構中以提供可以引入和篩選配體文庫的構建體。在一個實施方案中,捕獲樹脂的配體具有根據wo98/08603的公開內容中陳述的結構的結構。wo98/08603的捕獲樹脂包含合成的單環或雙環芳族或雜芳族配體以促進免疫球蛋白純化。wo98/08603涉及捕獲樹脂的結構的內容通過引用併入本文。wo98/08603描述了使用親和樹脂從細胞培養物上清液、血清、血漿或初乳中分離免疫球蛋白。在一個實施方案中,捕獲樹脂的配體具有根據wo2009/141384的公開內容中陳述的結構的結構。wo2009/41384的捕獲樹脂具有通式:其中r1、r2和r3代表分子量為15-1000g/mol、總重量為200-2000g/mol的有機部分,其配體通過r1、r2和r3之一通過醯胺鍵被固定化在固相支持物上。wo2009/141384涉及捕獲樹脂的結構的內容通過引用併入本文。wo2009/141384描述了配體結合蛋白質因子vii多肽。在一個實施方案中,捕獲樹脂的配體具有根據us20010045384的公開內容中陳述的結構的結構。us20010045384的捕獲樹脂為基於亞氨基二乙酸酯(ida)型支架組裝的蛋白a模擬物配體-複合物。us20010045384涉及捕獲樹脂的結構的內容通過引用併入本文。ida支架用三嗪基配體衍生化以提供多價三嗪基配體-複合物。在us20010045384中定義的說明性的三嗪基配體複合物在下面示出:已經證明該蛋白a模擬物用作用於免疫球蛋白諸如igg的親和純化介質。推斷配體-複合物中相鄰的三嗪基配體的分子幾何結構為使用ida支架的優點。us20010045384中定義的另一個說明性複合物在下面示出:該支鏈多價鄰苯二甲酸-配體支架蛋白a模擬物配體-複合物被證明對免疫球蛋白具有親和性。在一個實施方案中,捕獲樹脂的配體具有根據wo9710887和us6117996的公開內容中陳述的結構的結構。wo9710887和us6117996涉及捕獲樹脂的結構的內容通過引用併入本文。wo9710887和us6117996公開了以下類型的三嗪基-配體親和構建體:其中,(a)代表三嗪支架與多糖固體支持物的共價附接點,任選地通過在配體和固體支持物之間插入的間隔臂,並且r1和q為對蛋白質物質具有親和性的任選地取代的配體。有機部分被描述為蛋白a模擬物,並且被提出和示例為用於純化蛋白質物質的蛋白a的替代純化介質。在一個實施方案中,捕獲樹脂的配體具有根據wo2004/035199的公開內容中陳述的結構的結構。wo2004/035199涉及捕獲樹脂的結構的內容通過引用併入本文。wo2004/035199公開了包含以下通式的支鏈配體支架的蛋白l模擬物的用途,其中r1和r2相同或不同,並且各自為任選地取代的烷基或芳基配體,並且r3為任選地被間隔基序附接的固體支持物。三嗪基-配體支架已被公開為用於親和結合免疫球蛋白或其片段抗體(fab)的合適的蛋白l模擬物配體。此外,公開了這些三嗪基-配體支架對免疫球蛋白k和λ輕鏈具有優先結合親和性。在一個實施方案中,捕獲樹脂的配體具有根據us20110046353的公開內容中陳述的結構的結構。us20110046353涉及捕獲樹脂的結構的內容通過引用併入本文。us20110046353公開了片段抗體(fab)從產生介質的純化。不能在蛋白a介質上純化片段抗體。fab被表徵為具有能夠結合抗原的結合結構域,並且在其中公開的許多實施方案中,fab由一個重鏈(vh)或其功能片段和一個輕鏈(v1)或其功能片段連同至少一個其他鏈組成。其中定義了fab的親和配體,該親和配體由以下式的支鏈三嗪基支架組成,其中q代表對固體支持物基質的附接點,任選地具有間隔基序,並且基團a和b為被一個或更多個能夠氫鍵鍵合的取代基,優選地一個或更多個-oh、-sh或-co2h取代的苯基或萘基基團。已經報導使用以商品名mabsorbenta1p和mabsorbenta2ph和fabsorbentf1p從prometicbiosciences商購可得的支持的親和配體的優異結果。在一個實施方案中,捕獲樹脂的配體具有結構:在一個實施方案中,捕獲樹脂的配體具有結構:在一個實施方案中,捕獲樹脂的配體具有結構:在一個實施方案中,捕獲樹脂呈珠的形式。在一個實施方案中,在珠直徑方面,珠的尺寸為約10μm至約2000μm,優選地約50μm至約1000μm,並且最優選地約75μm至約500μm。在一個實施方案中,捕獲樹脂包括由選自由以下組成的組的物質製成的移動支持物:聚苯乙烯、聚苯乙烯(ps-dvb)-與二乙烯基苯(0.1-5.0%dvb,稱為微孔的(microporous))輕度交聯、聚苯乙烯(ps-dvb)-與二乙烯基苯(5-60%dvb,稱為大孔的(macroporous))高度交聯、聚乙二醇、聚乙二醇接枝的聚苯乙烯(ps-peg共聚物)、聚丙烯醯胺、可控孔度玻璃(controlledporeglass)(cpg)珠、二氧化矽(silica)、硅藻土、聚丙烯、聚四氟乙烯、聚乙烯、纖維素、聚甲基丙烯酸酯、官能化整料(functionalisedmonoliths)、官能化纖維、整體柱(monolithiccolumn)(諸如nikzad等人,oprd,2007,11,458-462)、官能化膜、瓊脂糖、瓊脂糖凝膠(sepharose)和磁性可回收聚合物珠。在優選的實施方案中,捕獲樹脂為由選自由以下組成的組的物質製成的移動支持物:瓊脂糖、瓊脂糖凝膠和纖維素。在一個實施方案中,捕獲樹脂為商購可得的捕獲樹脂,諸如fabsorbenttmf1phf樹脂。在一個實施方案中,捕獲樹脂為商購可得的捕獲樹脂,諸如mabsorbenttma1p或a2p樹脂。生物分子:在一個實施方案中,生物分子天然地存在於活生物體中。可選擇地,生物分子可以為天然地存在於活生物體中的化學化合物的衍生物。例如,生物分子可以為以不影響其生物活性的方式被化學地或遺傳地改變的生物分子。因此,在一個實施方案中,生物分子為重組生物分子,例如重組工程化的或重組修飾的生物分子。在一個實施方案中,生物分子為抗體。在一個實施方案中,生物分子為修飾的抗體,例如包括非天然胺基酸的抗體。在一個實施方案中,生物分子為抗體片段。在一個實施方案中,抗體為單克隆抗體。在一個實施方案中,抗體為曲妥珠單抗。在一個實施方案中,抗體、修飾的抗體或抗體片段為免疫球蛋白(ig),例如五種人免疫球蛋白種類之一:igg、iga、igm、igd和ige。術語抗體涵蓋單克隆抗體。術語抗體涵蓋多克隆抗體。術語抗體涵蓋抗體片段,只要它們顯示期望的生物活性。抗體可以為人抗體、動物抗體、鼠(murine)抗體、人源化抗體或包含人和動物序列的嵌合抗體。抗體結構的基本單元為至少20,000道爾頓例如約150,000道爾頓的異四聚體糖蛋白複合物。抗體長度可以為至少900個胺基酸,例如長度為1400個胺基酸。抗體可以包含兩個相同的輕(l)鏈和兩個相同的重(h)鏈,通過非共價締合和通過二硫鍵兩者連接在一起。每條重和輕鏈還具有規則間隔的鏈內二硫橋。每條重鏈為約50,000道爾頓。每條重鏈長度為至少300個胺基酸,例如長度為約450個胺基酸。抗體可以為僅有重鏈的抗體。每條輕鏈為約20,000道爾頓。每條輕鏈長度為至少100個胺基酸,例如長度為約250個胺基酸。抗體生物分子可以包含兩個相同的多肽鏈對,每對具有一條輕鏈和一條重鏈。每條輕鏈和重鏈依次由兩個區域組成:參與結合靶抗原的可變(「v」)區域和與免疫系統的其他組分相互作用的恆定(「c」)區域。輕鏈和重鏈可變區域在三維空間中一起形成結合抗原(例如,細胞表面上的受體)的可變區域。在一個實施方案中,生物分子為抗體片段。抗體片段包含全長抗體的一部分,通常為其抗原結合或可變區域。抗體片段的實例包括fab、pfc』、f(ab』)2和scfv片段;雙鏈抗體(diabodies);dsfv、線性抗體;親和抗體(affibody);微型抗體(minibody);單鏈抗體生物分子;包括納米抗體(nanobody)和可變新抗原受體(vnar)和由抗體片段形成的多特異性抗體。抗體片段長度可以為至少10個胺基酸,例如抗體片段長度可以為至少20個、40個、60個、80個、100個、120個、140個、160個、180個、200個、220個、240個、260個、280個或300個胺基酸。在一個實施方案中,生物分子為修飾的抗體或修飾的抗體片段。「修飾的抗體」或「修飾的抗體片段」意指由於至少一個胺基酸修飾而與親本抗體不同的抗體。修飾的抗體或修飾的抗體片段為在進行本發明的方法之前已經先前被化學或酶促修飾的或遺傳工程化的(例如,以包括非天然胺基酸等)抗體或抗體片段。修飾的抗體或修飾的抗體片段為指與野生型抗體相比,關於其尺寸不同或關於其糖基化不同但對其配體具有與野生型抗體相似的親和性的抗體。藥物:術語「藥物」包括當被施用到活生物體的體內時改變正常身體功能的任何物質。通常,藥物為用於治療、治癒、預防或診斷疾病或用於以其他方式增強身體或精神健康的物質。在一個實施方案中,藥物為細胞毒性藥物。到目前為止,領先的『超效力』(藥物)候選物被定義為兩類:(i)微管蛋白抑制物;和(ii)dna相互作用劑。微管蛋白抑制物調節微管蛋白聚合。dna相互作用劑靶向細胞dna。在一個實施方案中,藥物為微管蛋白抑制物。在一個實施方案中,微管蛋白抑制物選自由以下組成的組:(a)奧瑞他汀;和(b)美登素衍生物。在一個實施方案中,藥物為奧瑞他汀。奧瑞他汀包括天然存在的化合物多拉司他汀-10(dolastatin-10)的合成衍生物。奧瑞他汀為屬於抗腫瘤/細胞毒性假肽(pseudopeptide)家族。由於摻入了天然生物合成產物中鑑定出的4種非常規胺基酸(dolavaine、dolaisoleuine、dolaproine和dolaphenine),多拉司他汀在結構上為獨特的。另外,該類天然產物具有通過由pettit等人(us4,978,744)的總合成研究定義的許多不對稱中心。從結構活性關係看,dolaisoleuine和dolaproine殘基對於抗腫瘤活性似乎為必需的(us5,635,483和us5,780,588)。在一個實施方案中,奧瑞他汀選自由以下組成的組:奧瑞他汀e(ae);單甲基奧瑞他汀e(monomethylauristatine)(mmae);奧瑞他汀f(mmaf);vcmmae;和vcmmaf。在一個實施方案中,藥物為美登素或美登素的結構類似物。在一個實施方案中,藥物為美登素。美登素包括結構複雜的抗有絲分裂的聚酮化合物。美登素為導致腫瘤細胞凋亡的微管蛋白組裝的有效抑制物。在一個實施方案中,美登素選自由以下組成的組:mertansine(dm1);和美登素的結構類似物,諸如dm3或dm4。優選地,藥物為mertansine(dm1)。在一個實施方案中,藥物為dna相互作用劑。dna相互作用劑被稱為『超效力』(藥物)候選物。在一個實施方案中,dna相互作用劑選自由以下組成的組:(a)加利車黴素,(b)倍癌黴素和(c)吡咯並苯二氮卓類(pbd)。在一個實施方案中,藥物為加利車黴素。加利車黴素為引起雙鏈dna斷裂、導致細胞死亡的有效的細胞毒性劑。加利車黴素為天然存在的烯二炔(enediyne)抗生素(a.l.smith等人,j.med.chem.,1996,39,11,2103-2117)。在土壤微生物棘孢小單胞菌(micromonosporaechinospora)中發現加利車黴素。在一個實施方案中,加利車黴素為加利車黴素γ1。在一個實施方案中,藥物為倍癌黴素。倍癌黴素為有效的抗腫瘤抗生素,其通過在dna雙鏈體的小溝中選擇性地結合序列發揮其生物作用,並將腺嘌呤的n3烷基化(d.boger,pure&appl.chem.,1994,66,4,837-844)。在一個實施方案中,倍癌黴素選自由以下組成的組:倍癌黴素a;倍癌黴素b1;倍癌黴素b2;倍癌黴素c1;倍癌黴素c2;倍癌黴素d;倍癌黴素sa;環丙基苯並吲哚(cyclopropylbenzoindole)(cbi)倍癌黴素;centanamycin;瑞秋黴素(rachelmycin)(cc-1065);阿多來新(adozelesin);比折來新(bizelesin);和卡折來新(carzelesin)。在一個實施方案中,藥物為吡咯並苯二氮卓類。吡咯並苯二氮卓類(pbd)為一類天然存在的抗腫瘤抗生素。在鏈黴菌(streptomyces)中發現吡咯並苯二氮卓類。pbd通過在小溝中在嘌呤-鳥嘌呤-嘌呤單元處與dna特異性地共價結合來發揮其抗腫瘤活性。它們經由氨基鍵插入到鳥嘌呤的n2上,並且由於它們的形狀,其引起對dna螺旋的最小破壞。認為dna-pbd加合物的形成抑制核酸合成並引起dna螺旋中的切除依賴性單鏈和雙鏈斷裂。作為合成衍生物,兩個pbd單元經由柔性聚亞甲基系鏈(polymethylenetether)連接在一起允許pbd二聚體交聯相對的dna鏈,產生高度致死性損傷。在一個實施方案中,藥物為經由柔性聚亞甲基系鏈連接在一起的兩個吡咯並苯二氮卓類單元的合成衍生物。在一個實施方案中,吡咯並苯二氮卓類選自由以下組成的組:氨茴黴素(anthramycin)(及其二聚體);甲基氨茴黴素(mazethramycin)(及其二聚體);茅屋黴素(tomaymycin)(及其二聚體);prothracarcin(及其二聚體);芝加黴素(chicamycin)(及其二聚體);新絲拉黴素a(neothramycina)(及其二聚體);新絲拉黴素b(及其二聚體);dc-81(及其二聚體);西伯利亞黴素(sibiromycin)(及其二聚體);porothramycina(及其二聚體);porothramycinb(及其二聚體);西巴黴素(sibanomycin)(及其二聚體);abbeymycin(及其二聚體);sg2000;和sg2285。在一個實施方案中,藥物為通過烷基化靶向dna鏈間交聯的藥物。通過烷基化靶向dna鏈間交聯的藥物選自:dna靶向氮芥(dnatargetedmustard);鳥嘌呤特異性烷化劑;和腺嘌呤特異性烷化劑。在一個實施方案中,藥物為dna靶向氮芥。例如,dna靶向氮芥可以選自由以下組成的組:寡聚吡咯;寡咪唑(oligoimidazole);雙-(苯並咪唑)(bis-(benzimidazole))載體;聚苯甲醯胺(polybenzamide)載體;和9-苯胺基吖啶-4-甲醯胺(9-anilinoacridine-4-carboxamide)載體。在一個實施方案中,藥物選自由以下組成的組:紡錘菌素(netropsin);偏端黴素(distamycin);lexitropsin;他莫司汀(tallimustine);二溴他莫司汀(dibromotallimustine);pnu157977;和men10710。在一個實施方案中,藥物為雙-(苯並咪唑)載體。優選地,藥物為hoechst33258。鳥嘌呤特異性烷化劑為在特定核苷位置反應的高度區域特異性烷化劑。在一個實施方案中,藥物為選自由以下組成的組的鳥嘌呤特異性烷化劑:g-n2烷化劑(alkylator);a-n3烷化劑;絲裂黴素;carmethizole類似物;海鞘素類似物。在一個實施方案中,絲裂黴素選自:絲裂黴素a;絲裂黴素c;紫菜黴素(porfiromycin);和kw-2149。在一個實施方案中,carmethizole類似物選自:雙-(羥甲基)雙吡咯烷(bis-(hydroxymethyl)pyrrolizidine);和nsc602668。在一個實施方案中,海鞘素類似物為海鞘素743。腺嘌呤特異性烷化劑為在多聚嘧啶(polypyrimidine)序列中腺嘌呤的n3處反應的區域特異性和序列特異性小溝烷化劑。cyclopropaindolone和倍癌黴素可以被定義為腺嘌呤特異性烷化劑。在一個實施方案中,藥物為cyclopropaindolone類似物。優選地,藥物選自:阿多來新;和卡折來新。在一個實施方案中,藥物為苯基吲哚酮(benz[e]indolone)。優選地,藥物選自:cbi-tmi;和iso-cbi。在一個實施方案中,藥物為比折來新。在一個實施方案中,藥物為海洋抗腫瘤藥物(marineantitumordrug)。海洋抗腫瘤藥物一直為抗腫瘤藥物開發領域的發展領域(i.bhatnagar等人,mar.drugs2010,8,p2702-2720和t.l.simmons等人,mol.cancerther.2005,4(2),p333-342)。包括海綿、海綿-微生物共生群叢(association)、柳珊瑚(gorgonian)、放線菌和軟珊瑚的海洋生物已經被廣泛探索用於潛在的抗癌劑。在一個實施方案中,藥物選自:阿糖胞苷(cytarabine),ara-c;曲貝替定(trabectedin)(et-743);和甲磺酸艾日布林(eribulinmesylate)。在一個實施方案中,甲磺酸艾日布林選自:(e7389);soblidotin(tzt1027);乳酸角鯊胺(squalaminelactate);cemadotinplinabulin(npi-2358);plitidepsin;elisidepsin;zalypsis;tasidotin、synthadotin;(ilx-651);圓皮海綿內酯(discodermolide);ht1286;laf389;kahalalidef;krn7000;苔蘚抑素1(bryostatin1);hemiasterlin(e7974);marizomib;salinosporamidea;npi-0052);ly355703;crypto52;縮酚酸肽(depsipeptide)(nsc630176);海鞘素743;synthadotin;kahalalidef;角鯊胺(squalamine);脫氫膜海鞘素b(dehydrodidemninb);膜海鞘素b(didemninb);西馬多丁(cemadotin);soblidotin;e7389;nvp-laq824;圓皮海綿內酯;hti-286;laf-389;krn-7000(agelasphin衍生物);curacina;dmmc;salinosporamidea;laulimalide;vitilevuamide;含氯環肽(diazonamide);五加素(eleutherobin);匍枝珊瑚醇(sarcodictyin);pelorusidea;salicylihalimidesa和b;噻可拉林(thiocoraline);ascididemin;variolins;片螺素d(lamellarind;);dictyodendrins;es-285(spisulosine);和軟海綿素b(halichondrinb)。以下藥物也被本發明涵蓋:鵝膏菌屬(amanita)的擔子菌(basidiomycetes)產生的鵝膏毒蕈肽(α-鵝膏毒環肽)-二環八肽(bicyclicoctapeptides),例如青鬼尾鵝膏菌(greendeathcapmushroom);tubulysins;細胞溶素;dolabellanins;埃博黴素a、b、c、d、e、f。埃博黴素-構成一類非紫杉烷微管蛋白聚合劑,並通過粘細菌(myxobacterium)sorangiumcellulosum的天然發酵獲得。這些部分具有有效的細胞毒活性,這與微管的穩定有關,並導致在g2/m轉換期的有絲分裂停滯。埃博黴素已經跨越一組癌細胞系表現出有效的細胞毒性,並且通常表現出比紫杉醇(paclitaxel)更高的效力(x.:pivot等人,europeanoncology,2008;4(2),p42-45)。在一個實施方案中,藥物為鵝膏毒蕈肽。在一個實施方案中,藥物為tubulysin。在一個實施方案中,藥物為細胞溶素。在一個實施方案中,藥物為dolabellanin。在一個實施方案中,藥物為埃博黴素。以下藥物也被本發明涵蓋。在一個實施方案中,藥物選自:多柔比星(doxorubicin);表柔比星(epirubicin);依索比星(esorubicin);地託比星(detorubicin);嗎啉代-多柔比星;氨甲喋呤(methotrexate);甲氨喋呤(methopterin);博萊黴素(bleomycin);二氯甲氨喋呤(dichloromethotrexate);5-氟尿嘧啶;胞嘧啶-β-d-阿拉伯呋喃糖苷(cytosine-β-d-arabinofuranoside);紫杉酚(taxol);蛇形菌素(anguidine);美法侖(melphalan);長春花鹼(vinblastine);擬莖點黴毒素a(phomopsina);核糖體失活蛋白(rips);柔紅黴素(daunorubicin);長春花生物鹼(vincaalkaloids);伊達比星(idarubicin);美法侖;順鉑;蓖麻毒蛋白(ricin);肥皂草毒蛋白(saporin);蒽環類抗生素(anthracyclines);吲哚-苯二氮卓類(indolino-benzodiazepines);6-巰基嘌呤(6-mercaptopurine);放線菌素;環氧長春鹼(leurosine);異長春鹼(leurosideine);洋紅黴素(carminomycin);氨基喋呤(aminopterin);他利黴素(tallysomycin);足葉草毒素(podophyllotoxin);依託泊苷(etoposide);髮夾聚醯胺(hairpinpolyamides);磷酸依託泊苷;長春花鹼;長春花新鹼(vincristine);脫乙醯長春花鹼(vindesine);泰索帝視黃酸(taxotereretinoicacid);n8-乙醯基亞精胺(n8-acetylspermidine);喜樹鹼;埃斯培拉黴素(esperamicin);和烯二炔類(ene-diynes)。在一個實施方案中,藥物為多柔比星。在一個實施方案中,藥物為表柔比星。在一個實施方案中,藥物為依索比星。在一個實施方案中,藥物為地託比星。在一個實施方案中,藥物為嗎啉代-多柔比星。在一個實施方案中,藥物為氨甲喋呤。在一個實施方案中,藥物為甲氨喋呤。在一個實施方案中,藥物為博萊黴素。在一個實施方案中,藥物為二氯甲氨喋呤。在一個實施方案中,藥物為5-氟尿嘧啶。在一個實施方案中,藥物為胞嘧啶-β-d-阿拉伯呋喃糖苷。在一個實施方案中,藥物為紫杉酚。在一個實施方案中,藥物為蛇形菌素。在一個實施方案中,藥物為美法侖。在一個實施方案中,藥物為長春花鹼。在一個實施方案中,藥物為擬莖點黴毒素a。在一個實施方案中,藥物為核糖體失活蛋白(rip)。在一個實施方案中,藥物為柔紅黴素。在一個實施方案中,藥物為長春花生物鹼。在一個實施方案中,藥物為伊達比星。在一個實施方案中,藥物為美法侖。在一個實施方案中,藥物為順鉑。在一個實施方案中,藥物為蓖麻毒蛋白。在一個實施方案中,藥物為肥皂草毒蛋白。在一個實施方案中,藥物為蒽環類抗生素。在一個實施方案中,藥物為吲哚-苯二氮卓類。在一個實施方案中,藥物為6-巰基嘌呤。在一個實施方案中,藥物為放線菌素。在一個實施方案中,藥物為環氧長春鹼。在一個實施方案中,藥物為異長春鹼。在一個實施方案中,藥物為洋紅黴素。在一個實施方案中,藥物為氨基喋呤。在一個實施方案中,藥物為他利黴素。在一個實施方案中,藥物為足葉草毒素。在一個實施方案中,藥物為依託泊苷。在一個實施方案中,藥物為髮夾聚醯胺。在一個實施方案中,藥物為磷酸依託泊苷。在一個實施方案中,藥物為長春花鹼。在一個實施方案中,藥物為長春花新鹼。在一個實施方案中,藥物為脫乙醯長春花鹼。在一個實施方案中,藥物為泰索帝視黃酸。在一個實施方案中,藥物為n8-乙醯基亞精胺。在一個實施方案中,藥物為喜樹鹼。在一個實施方案中,藥物為埃斯培拉黴素。在一個實施方案中,藥物為烯二炔類。生物分子-藥物-綴合物:根據本發明,提供了通過本發明的方法可獲得的生物分子-藥物-綴合物。附圖簡述本發明的實施方案在下文中參考附圖被進一步描述,其中:圖1-通過實施例2產生的固相赫賽汀vcmmae綴合物的hic分析。從底部至頂部的跡線為赫賽汀-vce1.3、赫賽汀-vce2.4、赫賽汀-vce3.4、赫賽汀-vce4.4。洗脫曲線峰值為:rt4.3min-未綴合的赫賽汀,rt5.9min-藥物抗體比為2,rt7.5min-藥物抗體比為4,rt8.9min-藥物抗體比為6,和rt9.8min-藥物抗體比為8。圖2-通過實施例2產生的固相赫賽汀vcmmae綴合物的sec分析。從底部至頂部的跡線為赫賽汀、赫賽汀-vce1.3、赫賽汀-vce2.4、赫賽汀-vce3.4、赫賽汀-vce4.4。圖3-實施例3中產生的層析流動固相赫賽汀-vcmmae綴合物(chromatographicflowsolidphaseherceptin-vcmmaeconjugates)的hic分析。溶液相赫賽汀-vcmmae綴合物(上圖)、柱a製備的赫賽汀-vcmmae(中圖)、柱b製備的赫賽汀-vcmmae(下圖)的hic分析。圖4-實施例3中產生的層析流動固相赫賽汀-vcmmae綴合物的sec分析。溶液相赫賽汀-vcmmae綴合物(上圖)、柱a製備的赫賽汀-vcmmae(中圖)、柱b製備的-vcmmae(下圖)的sec分析。圖5-左側列顯示實施例5中在mabsorbenta1phftm樹脂上產生的赫賽汀-vcmmae綴合物的hic層析圖。右側列顯示同一赫賽汀-vcmmae綴合物的sec層析圖。層析數據表明,增加tcep與抗體的比增加了平均藥物抗體比(dar),並且隨著dar增加,使用固相技術未降低單體含量。圖6-實施例8中經由抗體的化學修飾和綴合在固相上合成的固相赫賽汀-vcmmae綴合物的hic分析。hic曲線表示隨機綴合中多種dar物類(0至8)特徵。圖7-經由實施例9中產生的固相工具合成的具有工程化半胱氨酸的赫賽汀-vcmmae綴合物。通過尺寸排阻層析(sec)分析綴合物以確定單體水平(上圖)。通過疏水相互作用層析(hic,中圖)和plrp(底圖)分析綴合物以計算藥物抗體比(dar)。圖8-實施例10中產生的-mcc-dm1和西妥昔單抗-mcc-dm1綴合物的sec跡線(樣品a至f)。通過使用『1步法』的固相技術合成的綴合物。在280nm處分析。詳述貫穿本說明書的描述和權利要求,詞語「包括(comprise)」和「包含(contain)」及它們的詞形變化意指「包括但不限於」,並且它們不意圖(並且不)排除其他部分、添加物、組分、整數或步驟。貫穿本說明書的描述和權利要求,單數涵蓋複數,除非上下文另有要求。具體地,在使用不定冠詞時,應理解本說明書預期多個以及單個,除非上下文另有要求。結合本發明的特定方面、實施方案或實例所描述的特徵、整數、特性、化合物、化學部分或基團應理解為可應用於本文所述的任何其他方面、實施方案或實例,除非與該方面、實施方案或實例不相容。在本說明書中公開的所有特徵(包括任何附隨的權利要求、摘要和附圖)和/或這樣公開的任何方法或過程的所有步驟可以以任何組合來組合,此類特徵和/或步驟中的至少某些相互排斥的組合除外。本發明並不被任何前述實施方案的細節限制。本發明擴展至在本說明書中公開特徵的任何新穎特徵或任何新穎組合(包括任何附隨的權利要求、摘要和附圖),或這樣公開的任何方法或過程的步驟的任何新穎的步驟或任何新穎的組合。讀者的注意被引導到與本說明書同時提交或在此之前提交的與本申請有關的、並且向公眾開放供查閱的所有論文和文獻,並且所有此類論文和文獻的內容通過引用併入本文。實施例在實施例中使用以下技術。尺寸排阻層析(sec)以0.5ml/min的流動速率在具有0.25mm氯化鉀和10%ipa的0.2m磷酸鉀ph6.95中使用tosohbiosciencetsk-gw3000swxl柱進行尺寸排阻層析。通過在280nm處的洗脫峰面積吸光度的積分確定綴合物的聚集狀態。疏水相互作用層析(hic)以0.8ml/min的流動速率在12分鐘內以緩衝液a至b的0-100%的線性梯度使用tosohtsk-丁基npr柱進行疏水相互作用層析。其中緩衝液a為具有25mm磷酸鈉的1.5m乙酸銨ph6.95,並且緩衝液b為具有25%ipa的25mm磷酸鈉ph6.95。通過在280nm處的洗脫峰面積吸光度的積分確定綴合物的抗體藥物比。反相層析(rp-plrp)以0.25ml/min的流動速率在31分鐘內以緩衝液a至b的25-95%的梯度使用agilentplrp-spl1912-1502柱進行反相(polymerlabsplrp)層析。其中緩衝液a為具有0.05%tfa的水,並且緩衝液b為具有0.04%tfa的acn。在注射前用包含50mmdtt的20mm硼酸鈉ph8.4在37℃下還原樣品15分鐘。通過在280nm處的洗脫峰面積吸光度的積分確定綴合物的抗體藥物比。通過uv分析的藥物抗體比對於uv分析,將樣品添加至具有1cm路徑長度的400ul石英比色杯中,並在thermoscientificmultiskango分光光度計上測量在252nm和280nm處的吸光度。基於公布的赫賽汀和dm1在這些波長處的摩爾消光係數,252nm和280nm數據用於計算藥物抗體比。實施例1-固相抗體藥物綴合物篩選該實施例證明,可以通過不需要方法開發的一般方法來將固定化的抗體與定義的藥物負載綴合。該方法適用於適應96孔板高通量篩選。通過將樹脂漿料和抗體溶液輕輕混合30分鐘,使赫賽汀(在pbsph7.4中,0.5ml,1mg/ml))與100μl(沉降樹脂體積)在pbs中平衡的fabsorbenttmf1phf樹脂結合。通過用pbs、2mmedta洗滌樹脂去除未結合的赫賽汀,並將樹脂最終重新懸浮於0.5mlpbs/edta中。通過將三(2-羧乙基)膦(tcep)鹽酸鹽添加至2mm的終濃度,並且然後將懸浮液在環境溫度下孵育18小時,使結合的赫賽汀(her)被還原。將樹脂用pbs/edta洗滌以去除未反應的tcep,並且然後重新懸浮於475μlpbs/edta中。添加vcmmae(vce)、n-乙基馬來醯亞胺(nem)和二甲基乙醯胺(dma)以實現1mm最終濃度的馬來醯亞胺(總vce和nem)和5%v/vdma。vce與nem的比在100:0、75:25、50:50、25:75和0:100間變化。通過將樹脂懸浮液在環境溫度下孵育60分鐘來綴合還原的抗體。將樹脂順序地用pbs/edta/5%v/vdma和0.1m甘氨酸ph5.0洗滌。用0.1m甘氨酸ph3.0洗脫綴合物。將洗脫的綴合物收集到2%v/v的1m三(羥甲基)氨基乙烷(tris)中以將其中和。然後通過尺寸排阻層析和反相層析(polymerlabs,plrp)層析分析中和的綴合物,以確定聚集體百分比和平均藥物載量。結果總結在下表1中:表1甚至最高負載藥物的綴合物的聚集體含量都可被接受用於抗原結合和基於細胞的測定中的進一步評價。用pbs/etda/5%v/vdma順序地洗滌,並且然後用0.1m甘氨酸ph5.0確保最終綴合物不含未反應的藥物接頭、nem和溶劑,並且不影響生物測定數據的解釋。用fabsorbenttmf1phf樹脂,該方法可用於篩選單克隆抗體組作為前導選擇的部分,以用於隨後抗體藥物綴合開發或用於從包含完整和fab片段抗體兩者的組織培養上清液直接產生adc。實施例2-批次模式(batchmode)下的固相部分tcep還原該實施例顯示可以通過部分還原鏈間二硫鍵隨後與vcmmae綴合來將固定化抗體與定義的藥物負載綴合,並且相對於在溶液中製備的相同綴合物,產品質量被增強。通過將樹脂漿料和抗體溶液輕輕混合30分鐘,使赫賽汀(0.5ml2mg/ml,pbs,ph7.4)與100μl(沉降樹脂體積)在pbs中平衡的fabsorbenttmf1phf樹脂結合。通過用pbs,2mmedta洗滌樹脂去除未結合的赫賽汀,並將樹脂最終重新懸浮於0.5mlpbs/edta中。通過將三(2-羧乙基)膦鹽酸鹽添加至每摩爾赫賽汀1至4摩爾tcep的比,並且然後將懸浮液在環境溫度下孵育2小時,使結合的赫賽汀被還原。添加vcmmae和二甲基乙醯胺(dma)以實現每摩爾赫賽汀2.5至10摩爾vcmmae和5%v/vdma,並且允許在環境溫度下進行綴合30分鐘。添加n-乙醯半胱氨酸(nac)以猝滅未反應的vcmmae並允許其反應20分鐘,然後順序地用pbs/edta/5%v/vdma和ph5.0的0.1m甘氨酸洗滌樹脂。將綴合物用0.1m甘氨酸ph3.0洗脫並收集到2%v/v的1m三(羥甲基)氨基乙烷(tris)中以使其中和。產生並分析對應系列的具有匹配的dar的赫賽汀與vcmmae的溶液相綴合物,以提供固相和溶液相綴合物質量的比較。然後通過疏水相互作用層析(圖1)和尺寸排阻層析(圖2)分析洗脫的綴合物,以確定聚集體百分比和平均藥物載量。結果總結在下表2中:表2數據顯示在固體支持物上tcep與抗體比與最終藥物載量之間的關係呈線性。另外,當與在溶液中製備的對應的綴合物相比時,固相綴合物顯示較低百分比的聚集體。實施例3-柱上的固相部分tcep還原該實施例顯示固定化的抗體綴合可以適用於具有優異再現性的層析流動過程。將赫賽汀(5ml2mg/ml,pbs,ph7.4)通過以120cm/小時負載與1ml的fabsorbenttmf1phf樹脂柱(先前在pbs中平衡)結合。通過用pbs、2mmedta平衡樹脂製備結合的赫賽汀用於還原。微型蠕動泵用於通過柱(在柱外部約200μl)創建小體積的pbs/edta再循環迴路,將tcep添加至柱以得到每摩爾赫賽汀2tcep的摩爾比。允許其在環境溫度下再循環120分鐘以還原赫賽汀。將儲存器(reservoir)和柱的內容物衝洗至廢棄物,並用pbs/edta/5%v/vdma代替,添加vcmmae,以得到每摩爾還原的赫賽汀5vcmmae的摩爾比。允許其在環境溫度下再循環60分鐘以綴合還原的赫賽汀。添加n-乙醯半胱氨酸(nac)以猝滅未反應的vcmmae並允許其反應20分鐘,然後順序地用pbs/edta/5%v/vdma和0.1m甘氨酸ph5.0洗滌樹脂。將綴合物用0.1m甘氨酸ph3.0洗脫並收集到2%v/v的1m三(羥甲基)氨基乙烷(tris)中以使其中和。使用第二柱/操作器在獨立的第二實驗中重複該過程。然後通過疏水相互作用層析(圖3)和尺寸排阻層析(圖4)分析洗脫的綴合物,以確定聚集體百分比和平均藥物載量。結果總結在下表3中:製備方法dar%聚集體赫賽汀00.2溶液相2.40.6柱a2.40.3柱b2.40.3表3數據顯示,當適用於層析流動模式時,vcmmae與赫賽汀的綴合與平均藥物載量、還原模式和聚集體產生一致。當tcep與抗體比匹配時,在批次模式和層析模式實現的dar為相同的。實施例4-批次模式下經由賴氨酸側鏈的smcc活化的赫賽汀與dm1固相綴合。該實施例顯示固定化抗體可以通過用smcc修飾而與賴氨酸的側鏈綴合,隨後與dm1綴合,並且相對於在溶液中製備的相同綴合物,產物質量被增強。通過將樹脂漿料和抗體溶液輕輕混合30分鐘,使赫賽汀(0.5ml4mg/ml,pbs,ph7.4)與100μl(沉降樹脂體積)在pbs中平衡的fabsorbenttmf1phf樹脂結合。通過用pbs,隨後用「修飾緩衝液」(50mmnapi、150mmnacl、2mmedtaph6.7)洗滌樹脂,去除未結合的赫賽汀,並將樹脂最終重新懸浮在包含5%v/vdma的修飾緩衝液中。通過添加4-(n-馬來醯亞胺基甲基)環己烷-1-羧酸琥珀醯亞胺酯(smcc)至每摩爾赫賽汀5至20摩爾smcc的比,並且然後將懸浮液在環境溫度下孵育2小時,使結合的赫賽汀被修飾。通過用pbs/5%v/vdma,隨後用『綴合緩衝液』(35mm檸檬酸鈉、150mmnacl、2mmedtaph5.0)洗滌樹脂,去除未反應的smcc,並將樹脂最終重新懸浮於包含3%v/vdma的綴合緩衝液中。添加dm1以實現每摩爾赫賽汀15摩爾dm1,並允許在環境溫度下進行綴合18小時。然後將樹脂順序地用pbs/edta/5%v/vdma和0.1m甘氨酸ph5.0洗滌。將綴合物用0.1m甘氨酸ph3.0洗脫並收集到2%v/v的1m三(羥甲基)氨基乙烷(tris)中以使其中和。通過使赫賽汀與7.6摩爾smcc,隨後每摩爾赫賽汀5摩爾dm1反應,產生具有約3.5的平均dar的赫賽汀-dm1溶液相綴合物,並分析其以提供固相和溶液相綴合物質量的比較。在修飾和綴合反應期間,赫賽汀的濃度分別為10mg/ml和5mg/ml。然後通過尺寸排阻層析和uv分析洗脫的綴合物以確定聚集體百分比和平均藥物載量。結果總結在下表4中:表4數據顯示在固體支持物上賴氨酸側鏈綴合為可能的,並且smcc與抗體比和最終載藥量之間的關係呈線性。另外,當與在溶液中製備的對應的綴合物相比時,儘管綴合反應期間蛋白濃度增加了四倍,但固相綴合物顯示出相當的聚集百分比。實施例5-比較蛋白a模擬物樹脂、蛋白l模擬物樹脂和傳統溶液相的批次模式下的固相部分tcep還原該實施例表明,在蛋白a模擬物樹脂或蛋白l模擬物樹脂上固定化抗體後可以實現抗體綴合。平行地採用使用相同的條件(出於比較目的)的傳統的溶液相方法。通過將樹脂漿料和抗體溶液輕輕混合30分鐘,使赫賽汀(0.5ml2mg/ml,pbs,ph7.4)與100μl(沉降的樹脂體積)在pbs中平衡的fabsorbenttma1phf和mabsorbenttma1phf樹脂兩者結合。通過用pbs、2mmedta洗滌樹脂去除未結合的赫賽汀,並將樹脂最終重新懸浮於0.5mlpbs/edta中。通過將三(2-羧乙基)膦鹽酸鹽添加至每摩爾赫賽汀1至4摩爾tcep的比,並且然後將懸浮液在環境溫度下孵育2小時,使結合的赫賽汀被還原。添加vcmmae和二甲基乙醯胺(dma)以實現每摩爾赫賽汀2.5至10摩爾vcmmae和5%v/vdma。允許在環境溫度下進行綴合15至30分鐘。添加n-乙醯半胱氨酸(nac)以猝滅未反應的vcmmae。在環境溫度下孵育20分鐘後,順序地用pbs/edta/5%v/vdma和0.1m甘氨酸ph5.0洗滌每種樹脂。將adc綴合物用0.1m甘氨酸ph3.0洗脫,並收集到2%v/v的1m三(羥甲基)氨基乙烷(tris)中以使其中和。對於溶液相反應,採用相同的還原和綴合條件,並且在比較分析之前經由g25批次脫鹽柱將最終的綴合物脫鹽到pbs中。所有綴合物均通過疏水相互作用層析(hic)和尺寸排阻層析(sec)被分析,以確定聚集體百分比和平均藥物載量(dar)。獲得的數據總結在下表5中:表5圖5中證明了使用mabsorbenttma1phf樹脂通過固相工具合成的赫賽汀-vcmmae綴合物的洗脫曲線。數據表明,可以在蛋白a模擬物樹脂或蛋白l模擬物樹脂上製備高質量的抗體藥物綴合物。另外,這些數據與較早的數據良好地一致(參見實施例2)。兩個數據集表明,當比較相同平均dar的綴合物時,通過固相方法合成的綴合物比通過類似溶液相方法合成的綴合物具有高的單體含量。實施例6-柱上抗體藥物綴合物的固相綴合的可擴展性流動模式合成對大規模製備具有吸引力。該實施例證明,綴合物的固相合成在多種柱尺寸的產品質量和產量方面為可擴展的和一致的。進行了一系列實驗以開發綴合過程,所述過程使用在柱/流動模式中適於fabsorbenttmf1phf樹脂的曲妥珠單抗/tcep/vcmmae綴合模式實現3.6±0.2的dar。然後通過增加柱直徑、柱長度和蛋白載量(mg/ml)來擴大模式,同時維持因素諸如線性流動速率、tcep/vcmmae與抗體比和反應時間。在小規模下通過g25緩衝液交換(pd10或hiprepxk16/10)配製釋放的綴合物,並且在更大規模下通過tff滲濾到在ph6的5mm組氨酸、50mm海藻糖、0.01%吐溫20配製釋放的綴合物。—有關測試的多種條件的概述,參見表6。通過用在ph7.5的柱運行緩衝液10mmtris/2mmedta洗滌,製備用於柱包裝(columnpacking)的fabsorbenttmf1phf樹脂。將柱以10cm/min包裝為50%漿料。使用相對於最終要求床體積的10%過量以允許在包裝期間樹脂壓縮。以2cm/min的固定線性流動速率進行所有負載、洗滌、反應和洗脫步驟。以24.1mg/ml的濃度供給曲妥珠單抗抗體。將曲妥珠單抗稀釋至2mg/ml,並在ph7.5在10mmtris/2mmedta緩衝液中將其負載到fabsorbenttmf1phf樹脂上,以實現要求的樹脂載量。在負載抗體之後,用5倍柱體積(cv)的在ph7.5的10mmtris/2mmedta緩衝液洗滌柱子。載量突破(loadbreakthrough)的uv分析和隨後的洗滌確認在所有靶載量下曲妥珠單抗的完全結合。使用微型蠕動泵和主柱頂部和底部的三通閥建立主柱外部的反應物儲存器/再循環迴路。調節儲存器體積以實現相對於主柱的50%的體積,並且這為所有過程反應物被加入的地方。通過將tcep(相對於曲妥珠單抗2.24當量)添加至儲存器中並在環境溫度下再循環2小時來實現二硫化物還原。用5cv的在ph7.5的10mmtris/2mmedta/5%dma緩衝液洗滌還原的曲妥珠單抗。通過將dma(5當量)中的10mmvcmmae添加至儲存器中並將這在環境溫度下再循環總計60分鐘來開始綴合。通過將n-乙醯半胱氨酸(nac,10當量)添加至儲存器來猝滅未反應的vcmmae。將所得混合物再循環20分鐘,然後進行最後的洗滌步驟。用5cv的在ph7.5的10mmtris/2mmedta/5%dma緩衝液,隨後用5cv的在ph7.5的10mmtris/2mmedta洗滌柱子,並且然後用10cv的具有0.1m甘氨酸ph3的步驟洗脫液洗脫。使用uv光譜分析確定含蛋白的級分,然後取決於規模將含蛋白的級分合併用於經由g25或tff的最終的緩衝液交換。將所有adc配製為在ph6的5mm組氨酸,50mm海藻糖、0.01%吐溫20並且稀釋至1mg/ml。hic、sec和rp-hplc層析方法用於確定平均dar、還原模式和最終配製後的單體含量。在g25或tff之前,對混合的釋放的級分通過rp-hplc進行剩餘的溶劑和剩餘的vcmmae的定量。作為直接比較,使用曲妥珠單抗和vcmmae以相似的方式進行三種溶液相綴合。使用500mm硼酸鹽、25mmedta將曲妥珠單抗的ph調節至ph8.2。通過在20℃下將曲妥珠單抗與tcep(相對於抗體1.94當量)孵育90分鐘來實現二硫化物的部分還原。在20℃下在30分鐘內完成還原的曲妥珠單抗與vcmmae(4.85當量)的綴合。然後在環境溫度下在20分鐘內用nac(4.85當量)猝滅過量的vcmmae。然後使用用於較小規模固相綴合的相同g25柱/過程來純化/配製綴合物,以提供具有3.6±0.2的靶dar的所得曲妥珠單抗-vcmmae綴合物。表6中的數據表明溶液相和固相合成兩者均可以一致地實現靶dar。然而,當與在多種規模的固相合成產生的數據直接比較時,來自溶液相的綴合產物的質量較差。溶液相方法遞送具有可測量水平的游離毒素接頭和溶劑兩者的綴合物。剩餘的毒素接頭將需要通過進一步純化被去除,然後綴合物可以用於任何體外細胞測定。表6中的數據還突出溶液相和固相綴合之間的重要區別。固相方法一致地遞送具有不可檢測水平的游離毒素和游離溶劑的綴合物。實質上,固相技術通過洗滌綴合物而純化掉多餘的試劑,同時將綴合物固定化在固相樹脂上以遞送優異純度的綴合物。下表6比較和對比通過固相和溶液相方法合成的曲妥珠單抗-vcmmae綴合物的合成。表6*1[dma]為%體積/體積,lod=<0.0001%v/v*2[毒素]=表示為游離和結合百分比的游離毒素,lod=<0.1%在ides/endoh/dtt處理後的ms分析在用ides(fabricatortm,genovis)、removeittmendos(newenglandbiolabs)和dtt處理後,通過ms分析來自表6的選擇的綴合物樣品。然後將重疊的離子系列解卷積,以提供由裸露的和綴合的(毒素)抗體片段組成的片段質量模式,從其可以使用信號強度計算通過ms確定的dar。收集的數據顯示在下表7中。表7使用該ms技術計算的平均dar跨越用於合成曲妥珠單抗-vcmmae綴合物的多種固相規模為一致的。此外,通過ms確定的dar和通過hic確定的dar非常一致。如通過對裸露的與綴合的lc和fd片段的一致響應分數示出的,還原的模式為一致的。該分析突出溶液和固相綴合之間的還原的模式的微妙差異。如通過固相和溶液的不同的平均lc/lc+vce比示出的,在溶液中,在hl二硫化物處存在更多的還原。綴合物的細胞殺傷分析在抗原陽性細胞殺傷測定中,分析來自表6的選擇的綴合物樣品的效力。將sk-br3細胞用胰蛋白酶/edta收穫,並且然後在測定介質中洗滌,並且然後用更多的測定介質稀釋至0.9×105/ml。將100μl該細胞儲備液添加至96孔板的每一個孔中,並將板在37℃/5%co2下孵育3小時以使細胞定居。在測定介質中酌情稀釋樣品和標準品,並酌情將100μl添加至孔中。將細胞/樣品孵育72小時,並且然後使用商業ldh測定試劑盒測量%細胞細胞毒性。在sk-br3her2陽性細胞殺傷測定(ldh測定)中,無論是由固相樹脂還是溶液中製成,綴合物為等效的。數據總結在下表8中。表8測定1以一式兩份運行樣品。測定2以一式三份運行樣品。表8中的平均欄為來自測定1和2的所有5個數據點的平均值。結果表明,在抗原陽性細胞殺傷測定中,通過固相或溶液相工具合成的adc綴合物之間的adc療效無差異。實施例7-赫賽汀與用smcc預活化的dm1固相綴合該實施例表明,通過用預活化的dm1-smcc細胞毒素藥物接頭修飾可以將固定化的抗體綴合在賴氨酸的側鏈上。該方法被稱為產生綴合物的「1步法』。通過將過量的dm1-sh與smcc一起孵育以驅動偶聯反應完成,並且然後使用該粗製混合物進行綴合來製備活化的dm1-smcc。在環境溫度下在5小時內,在dma中用異雙功能交聯劑smcc(相對於smcc的1.6當量的dm1)將巰基官能化的細胞毒素dm1預活化。基於dm1至dm1-smcc的100%轉化確定理論dm1-smcc濃度。將fabsorbenttmf1phf樹脂(100μl)用1或2mg的赫賽汀負載,並將其懸浮於包含在ph6.7的50mmnapi、150mmnacl、2mmedta的修飾緩衝液(360μl)中。將dm1-smcc藥物接頭連同dma一起添加至漿料中,至終濃度為10%v/v。使用三種不同的dm1-smcc過量:相對於mab結合的5、10和15當量。在環境溫度下,將綴合反應在旋轉器上攪拌2小時。綴合後去除上清液,並將樹脂順序地用pbs中的10%v/vdma,隨後單獨的pbs緩衝液ph7.4進行洗滌。用0.1m琥珀酸ph2.6從樹脂上切下綴合物(2x500μl,在環境溫度下孵育5分鐘)。通過uv和sec分析來分析所得綴合物,以確定平均dar和單體含量。結果總結在下表9中。表9觀察到使用的dar與dm1-smcc過量之間的線性關係,其中dm1-smcc的增加導致dar的增加。還可以通過增加赫賽汀至固相樹脂上的載量來增加dar。無論dar如何,單體水平高,所有綴合物均高於99.5%。實施例8-化學修飾的抗體用於抗體藥物綴合物的固相合成的應用該實施例表明使用化學修飾的抗體結合固相綴合技術的adc的合成。抗體首先在溶液中被化學還原,然後被孵育並結合至固相樹脂,之後綴合過程發生在樹脂上。在環境溫度下在90分鐘持續時間內用還原劑1mmtcep(相對於抗體2當量)還原赫賽汀抗體(1ml1mg/ml,在pbs中,ph7.4,2mmedta)。用4×等份的50mmnapi,ph8緩衝液洗滌fabsorbenttmf1phf樹脂(100μl)。去除過量的緩衝液,提供潮溼的樹脂,在其中裝載1ml在pbs,ph7.4,2mmedta中的還原的赫賽汀。在環境溫度下,將所得抗體樹脂漿料在旋轉器上攪拌30分鐘。然後將漿料離心,去除上清液,然後通過uv分析上清液以通過減去吸光度確定抗體結合。然後用1mlpbs,ph7.4,2mmedta緩衝液洗滌抗體-樹脂。還分析洗滌級分以確認與樹脂結合的抗體的總體濃度。將還原的抗體樹脂懸浮於pbsph7.4,2mmedta緩衝液(950μl)和dma(4.6μl)中。將dma(10mm,3.3μl)中的細胞毒性藥物接頭mcf或vcmmae裝載至樹脂漿料中,以提供在pbsph7.4,2mmedta介質中的總體5%v/vdma。將綴合反應在環境溫度下進行30分鐘,同時在旋轉器上輕輕攪拌。通過將nac(10mm,3.3μl)添加至漿料中猝滅綴合反應,並將所得混合物在環境溫度下輕輕攪拌20分鐘。然後將樹脂過濾並用在pbs,ph7.4的中的2×5%v/vdma洗滌。通過在環境溫度下用0.1m甘氨酸,ph3(980μl)處理5分鐘,從固相樹脂中釋放adc綴合物。然後將樹脂漿料離心並取出上清液。將單次裝載(singlecharge)的20ml的1mtris緩衝液添加至上清液中,以製備適於uv分析的1ml樣品,以確定回收產量。然後直接對樣品進行分析;然後通過疏水相互作用層析(hic)在g25包裝的柱上脫鹽,以計算藥物抗體比(dar)。圖6顯示了來自該固相綴合的dar物類的展寬。數據計算出在280nm處歸一化的平均2.2的dar。圖6中的hic曲線為通過溶液相或固相的隨機綴合的特徵。實施例9-使用重組工程化硫醇抗體的固相位點特異性綴合該實施例示例了使用固相樹脂與重組工程化硫醇抗體合成抗體藥物綴合物。在該實施例中,重組工程化抗體包含2個另外的半胱氨酸殘基,其促進與thiomab抗體技術(genentech)相似的位點特異性綴合技術。在包含5mm組氨酸、50mm海藻糖和0.01%v/vps20(濃度為20mg/ml)的製劑緩衝液中供給1ml具有工程化半胱氨酸(v205ckabat編號)的赫賽汀溶液。用4ml10mmtris,ph7.5緩衝液稀釋1ml抗體溶液。將得到的抗體溶液與在10mmtrisph7.5緩衝液中預平衡的fabsorbenttmf1phf樹脂(1ml,沉降樹脂體積)一起孵育。使抗體結合至樹脂,將漿料在環境溫度下用旋轉器輕輕旋轉10分鐘。具有工程化半胱氨酸的結合的赫賽汀通過用大量過量的還原劑dtt(相對於抗體20當量)處理而完全被還原。在環境溫度下將還原懸浮液在旋轉器上輕輕攪拌16小時。然後用2×5ml50mmtris2.5mmedta,ph8緩衝液,隨後用2×5ml10mmtrisph7.5緩衝液洗滌樹脂以去除所有痕量的dtt。將樹脂最終重新懸浮於5ml的10mmtris,ph7.5緩衝液中。然後通過添加在dma中的脫氫抗壞血酸(dhaa)(相對於抗體10當量)將具有工程化半胱氨酸的固定化的抗體再氧化,並且在環境溫度下在1小時內將樹脂漿料在旋轉器上輕輕攪拌。然後將dma中的一次裝載的細胞毒性藥物接頭vcmmae(相對於抗體2.5當量)添加至樹脂上的具有工程化半胱氨酸的固定化的抗體中,以實現緩衝液混合物中5%v/vdma的最終組成。將綴合在環境溫度下伴隨旋轉進行1小時。然後通過用3×5ml在10mmtris,ph7.5緩衝液中的5%v/vdma,然後3×5ml10mmtris,ph7.5緩衝液洗滌,從樹脂漿料中去除過量的vcmmae。通過與0.1m甘氨酸,ph2.96(5ml)伴隨旋轉孵育10分鐘,從樹脂釋放adc綴合物。然後將釋放的赫賽汀與工程化半胱氨酸-vcmmae綴合物經由hightrap脫鹽柱(gehealthcare)立即配製到5mm組氨酸、50mm海藻糖、0.01%v/vps20,ph6的緩衝液中。然後通過尺寸排阻層析(sec)分析釋放的綴合物樣品以確定單體水平。疏水相互作用層析(hic)和rp-plrp用於計算藥物抗體比(dar)。這些分析的數據被報告在圖7中。分析性sec層析表明固相技術結合重組修飾的抗體提供了極好純度的adc綴合物,這通過非常高的單體含量證明。hic曲線還證明平均2.1的dar,並且0、1、2和更高的dar的分布與thiomabs的公布的結果一致。這表明固相可以用於產生在工程化抗體中摻入工程化半胱氨酸殘基的位點特異性adc。實施例10-固相和西妥昔單抗與dm1-smcc綴合該實施例表明『1步法』中抗體藥物綴合物的合成。為曲妥珠單抗(herceptin,roche)的生物仿製(biosimilar)單克隆抗體。本文我們證明了抗體藥物綴合物-dm1可以經由使用通過初審(pre-qualified)的活化的dm1毒素-接頭mcc-dm1使用固相綴合方法來合成。通過用pbs,ph7.4緩衝液稀釋,用儲備液樣品(25.6mg/ml)製備在2mg/ml、4mg/ml和6mg/ml的三種濃度的平行地,還通過用pbs,ph7.4緩衝液稀釋,用儲備液樣品(15.5mg/ml)製備2mg/ml、4mg/ml和6mg/ml的三種濃度的西妥昔單抗在280nm和320nm進行每一個樣品的uv吸收測量。用4×50μl等份的50mmnapi,ph8緩衝液洗滌fabsorbenttmf1phf樹脂(100μl)的六個單獨樣品,並去除上清液。每一個抗體濃度的0.5ml等份在1小時時間段內在旋轉器上伴隨輕輕攪動在環境溫度下與樹脂一起孵育。單獨地,將每一個樹脂樣品離心並取出上清液。每一個上清液通過uv在280nm和320nm處分析。抗體在每一個樹脂上的載量通過減去uv確定。然後用1×等份的pbs洗滌每一個樹脂以去除任何多餘的抗體。在三個單獨的ph下製備綴合緩衝液:0.1mnapi,ph7.5;0.1mnapi,ph8和0.1mnahco3,ph8.5。將具有結合抗體的樹脂樣品與每一種緩衝液單獨孵育。向每一個漿料中裝載dma中的毒素-接頭mcc-dm1。使用相對於抗體5至10當量的mcc-dm1的一系列過量當量。總體而言,mcc-dm1的裝載提供緩衝液中5%v/vdma的綴合介質。伴隨在旋轉器上輕輕攪拌在環境溫度下在2小時內進行綴合反應。在該持續時間之後,去除上清液,並且每一個樹脂用4×50μl等份的在pbsph7.4中的5%v/vdma,隨後用3×50μl等份的pbsph7.4進行洗滌。通過在環境溫度下在30分鐘內在50μl的在0.1m甘氨酸ph3中的50%v/v丙二醇中孵育每一個樹脂,從固相樹脂中取出抗體藥物綴合物。將上清液單獨地收集,並在214nm通過sec對其分析,以確定產量和單體含量。在252nm和280nm的sec分析促進dar的計算。表10中顯示從固相樹脂中取出綴合物(洗脫的)後即刻的綴合物數據。表10然後將所有綴合物脫鹽到製劑緩衝液中。對於綴合物,採用5mm組氨酸、50mm海藻糖、0.01%ps20,ph6的製劑緩衝液。對於西妥昔單抗,採用20mm琥珀酸鹽、150mmnacl、2mmedta,ph5.5的製劑緩衝液。所有綴合物均使用nap5柱脫鹽。然後通過sec層析重新分析脫鹽後樣品以確定最終物質的單體純度。製劑緩衝液中最終(配製的)綴合物的數據顯示於表10中。在圖8中證明-mcc-dm1和西妥昔單抗-mcc-dm1綴合物的sec跡線,並且指示高單體含量。數據清楚地表明,可以使用固相綴合技術實現始終如一的高單體產物。通過改變毒素-接頭過量,可以容易地實現不同dar的一系列綴合物。當前第1頁12