肟酯類化合物及其合成方法及應用與流程
2023-10-11 00:21:54 3
本發明涉及肟酯類化合物,尤其涉及一種用於UV固化材料的光引發劑及其製備方法。
背景技術:
紫外光(UV)固化,簡稱光固化技術,光固化是利用紫外光照射具有化學活性的液態材料,引發其快速聚合交聯,並使其瞬間固化的過程。
固化技術在20世紀80年代就被譽為是一項面向21世紀的綠色工業新技術,該技術是一種高效、環保、節能、優質的材料表面處理技術,在光固化塗料、光刻膠、光固化油墨、微電子、粘和劑、光碟複製、紙張上光技術中得到廣泛的應用,在光固化技術進步過程中,光引發劑體系的研究與開發始終佔據著十分重要的位置。
1968年Bayer公司開發了第一代紫外光固化木器塗料,首先實現了光固化技術的產業化。隨後光固化技術迅猛發展,應用領域不斷擴大,形成了一個新的產業。上世紀70、80年代,歐美輻射固化協會成立,推動了光固化技術的研究和發展,在北美、歐洲和日本等發達國家和地區,巴斯夫、拜耳、陶氏等跨國公司紛紛加盟光固化生產,目前已成為了具有一定市場規模的產業。我國從上世紀80年代開始發展光固化技術,由於原料和設備的限制,發展緩慢。進入90年代,紫外光固化技術和設備的引進大大推動了我國光固化產業的發展,進入21世紀,我國光固化產業獲得了更加快速的發展,特別是光引發劑已成為世界上最大的生產和出口國,已初步形成一個新的高新產業。如今大力提倡可持續發展,建立和諧社會,並加大了環境的保護,這為我國光固化產業的發展提供了機遇。
當前,關於光引發劑己經有了非常多的報導,例如安息香衍生物、聯苯醯縮酮類、α,α一二烷氧基苯乙酮類、二苯甲酮/胺類、米氏酮、噻唑酮/胺類、芳香重氮鹽、三氮嗪類、肟酯類等,代表例有可商購的Irgacure369、Darocurel173、OXE-l和OXE-2。然而,這些光引發劑在實際應用中均或多或少地存在感光度低(聚合速率和曝光劑量高)、溶解性差(透明度和光刻殘渣多)、貯存穩定性低、短波長靈敏度不足、對可光聚合單體的特定選擇性不強等缺陷,從而在整體上影響了感光材料的性能。
作為影響光固化組合物感光性能的最主要因素,光引發劑對可光聚合組合物感光性能的影響不僅體現在光引發劑本身對於輻射的敏感性,還在於它與可光聚合單體(或其組合物)之間的適配性。因此,在確定一種可光聚合組合物的配方時,最優的是尋找一種與內含的可聚合單體之間具有良好效應(例如,協同效應)的光引發劑,從而可以進一步優化組合物的感光性能。
作為優選的可光聚合組合物,一方面要求具有優異的儲存穩定性能;另一方面要對短波i 線和g線具有高靈敏度,以便降低曝光劑量,提高曝光效率,從而縮短生產周期;再一方面要求由光聚合組合物製備的光固化產品要圖像圖案精細整,沒有缺陷和浮渣,且光固化膜的硬度要好。這些指標對濾光片、光致抗蝕劑的性能尤為重要,而要達到這個效果,首先就要求組合物中使用的光引發劑本身在短波處要具有非常高的感光度,另外整個組合物體系要搭配的非常合理,即組合物中的可聚合成分(單體、樹脂)與體系中光引發劑配合使用時能發揮最佳應。目前,針對這方面的研究還非常的少。
現在入們對於光引發劑的效果及其分解產物的毒性、氣味和遷移性等特性要求越來越高,開發具有良好的溶解性、低氣味或無氣味和低遷移性的良好特性的大分子光引發劑將成為未來發展的主要方向。但目前已商業化的大分子光引發劑大多價格昂貴或產品性能有一定缺陷,因此迫切需要價格低廉且性能好的產品來替代。
技術實現要素:
發明目的:為了克服背景技術中現有醯基肟酯類光引發劑應用性能的不足,本發明的目的是提供一種溶解性好、熱穩定性好、反應活性高、生產成本低、價格低廉、基本無氣味,低遷移性,且使用安全性高(毒性低)的醯基肟酯類化合物。
本發明的另一目的是提供所述醯基肟酯類化合物的製備方法。
本發明的另一目的是提供所述醯基肟酯類化合物在UV光固化材料中的應用。
技術方案:為了達到上述發明目的,本發明提供了一種醯基肟酯類化合物,所述醯基肟酯類光化合物具有通式Ⅰ所示結構:
其中,
所述R1選自其中,所述X1為-S-、-S-S-、-O-、-NR-,其中R為氫、1-8個碳原子的烷烴基或烷氧基、3-8個碳原子的環烷基;所述X『和Y』相同或不同,各自獨立地選自-S-、-S-S-、-O-、-CO-;所述環狀結構中一個或多個-H可以被-F、-Cl、-Br、-I、-NO2、-COOR′1、-CONR′2、-OR′3、1-8個碳原子的烷基或烷氧基或2-8個碳原子的烯烴基取代,其中R』1和R』2各自獨立地選自-H、 1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基,R』3選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基或1-8個碳原子的碳鏈羰基,其中,1-8個碳原子的碳鏈羰基中羰基在端位,位於連接鍵處;
所述R3表示其中,所述X2為-S-、-S-S-、-O-、-NR-,其中R為氫、1-8個碳原子的烷烴基或烷氧基、3-8個碳原子的環烷基;所述環狀結構中一個或多個-H可以被-F、-Cl、-Br、-I、-NO2、-COOR′1、-CONR′2、-OR′3、1-8個碳原子的烷基或烷氧基或2-8個碳原子的烯烴基取代,其中R』1和R』2各自獨立地選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基,R』3選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基或1-8個碳原子的碳鏈羰基,其中,1-8個碳原子的碳鏈羰基中羰基在端位,位於連接鍵處;
所述R2選自-H、1-20個碳原子的烷基或烷氧基、3-8個碳原子的環烷基、2-20個碳原子的烯烴基或R1所示基團。
在本發明的一些實施方式中,優選地,所述R1選自其中,所述X1為-S-、-S-S-或-O-,其中,所述X『和Y』相同或不同,各自獨立地選自-S-、-O-或-CO-,所述環狀結構中一個或多個-H可以被-F、-Cl、-NO2、-COOR′1、-CONR′2、-OR′3、1-8個碳原子的烷基或烷氧基取代,其中R』1和R』2各自獨立地選自-H、1-8個碳原子的烷基,R』3選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基。
在本發明的一些實施方式中,優選地,所述R1選自
本發明的一些實施方式中,所述R3表示其中,所述X2為-S-、-S-S-、-O-、-NR-,其中,所述R為1-5個碳原子的烷烴基或烷氧基、3-8個碳原子的環烷基,所述環狀結構中一個或多個-H可以被-F、-Cl、-NO2、-COOR′1、-CONR′2、-OR′3、1-5個碳原子的烷基或烷氧基取代,其中R』1和R』2各自獨立地選自-H、1-8個碳原子的烷基,R』3選自-H、1-5個碳原子的烷基。
本發明的一些實施方式中,所述R3表示其中,所述X2為-S-、-S-S-、-O-、-NR-,其中,所述R為1-5個碳原子的烷烴基或烷氧基、3-8個碳原子的環烷基,所述環狀結構中一個或多個-H可以被-F、-Cl、-NO2、-COOR′1、-CONR′2、1-5個碳原子的烷基或烷氧基取代,其中R』1和R』2各自獨立地選自-H、1-8個碳原子的烷基。
本發明的一些實施方式中,所述X2選自-S-、-O-、-S-S-或-NR-,其中,所述R為1-5個碳原子的烷烴基或烷氧基、3-6個碳原子的環烷基。
本發明的一些實施方式中,優選地,所述R3優選自
本發明的一些實施方式中,優選地,所述R2選自-H、1-20個碳原子的烷基或烷氧基、3-8個碳原子的環烷基、2-20個碳原子的烯烴基、
本發明的一些實施方式中,優選地,所述R2選自-H、1-15個碳原子的烷基或烷氧基、3-8個碳原子環烷基、2-15個碳原子的烯烴基、
本發明的一些實施方式中,優選地,所述R2選自-H、1-15個碳原子的烷基或烷氧基、3-8個碳原子環烷基、2-15個碳原子的烯烴基、
本發明的一些實施方式中,優選地,所述R2選自1-15個碳原子的烷基或烷氧基、3-8個碳原子環烷基、2-15個碳原子的烯烴基、
本發明的一些實施方式中,通式Ⅰ的化合物選自由通式Ⅰ-1至Ⅰ-10所示化合物組成的組:
其中,
所述R2選自-H、1-20個碳原子的烷基或烷氧基、3-8個碳原子的環烷基、2-20個碳原子的烯烴基或R1所示基團。
本發明還提供了一種通式Ⅰ所述的醯基肟酯類化合物的製備方法,包含如下步驟:
a、中間體Ⅰ-A的合成:以苯、二苯硫醚或硫雜蒽酮等為起始原料,與含有R2基團的醯滷化合物,在三氯化鐵、三氯化鋁或氯化鋅等作用下,通過付克醯基化反應,合成中間體Ⅰ-A:
b、中間體Ⅰ-B的合成:中間體I-A在通有氯化氫或加鹽酸的情況下與亞硝酸甲酯、亞硝酸乙酯或亞硝酸異戊酯等進行氧化反應,生成醯基肟中間體Ⅰ-B:
c、醯基肟酯類光引發劑合成:中間體Ⅰ-B與含有M1結構的醯滷或酸酐,在吡啶或三乙胺等縛酸劑存在下,在二氯甲烷、二氯乙烷或二氧六環等做溶劑下合成通式Ⅰ的化合物。
其中,
所述R1選自其中,所述X1為-S-、-S-S-、-O-、-NR-,其中,所述R為氫、1-8個碳原子的烷烴基或烷氧基、3-8個碳原子的環烷基,所述X『和Y』相同或不同,各自獨立地選自-S-、-S-S-、-O-、-CO-,所述環狀結構中一個或多個-H可以被-F、-Cl、-Br、-I、-NO2、-COOR′1、-CONR′2、-OR′3、1-8個碳原子的烷基或烷氧基或2-8個碳原子的烯烴基取代,其中R』1和R』2各自獨立地選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基,R』3選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基或1-8個碳原子的碳鏈羰基,其中,1-8個碳原子的碳鏈羰基中羰基在端位,位於連接鍵處;
所述R3表示其中,所述X2為-S-、-S-S-、-O-、-NR-,其中,所述R為氫、1-8個碳原子的烷烴基或烷氧基、3-8個碳原子的環烷基,所述環狀結構中一個或多個-H可以被-F、-Cl、-Br、-I、-NO2、-COOR′1、-CONR′2、-OR′3、1-8個碳原子的烷基或烷氧基或2-8個碳原子的烯烴基取代,其中R』1和R』2各自獨立地選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基,R』3選自-H、1-8個碳原子的烷基、3-8個碳原子的環烷基、2-8個碳原子的烯烴基或1-8個碳原子的碳鏈羰基,其中,1-8個碳原子的碳鏈羰基中羰基在端位,位於連接鍵處;
所述R2選自-H、1-20個碳原子的烷基或烷氧基、3-8個碳原子的環烷基、2-20個碳原子的烯烴基或R1所示基團。
具體反應路線如下:
本發明中所述的步驟a中間體Ⅰ-A合成的具體操作為:氮氣保護下,向有機溶劑A中加入起始原料(苯、二苯硫醚或硫雜蒽酮)、AlCl3攪拌混合,冰鹽水浴冷卻至-5℃左右,滴加R2基團的醯滷化合物與有機溶液A的混合液,溫度控制在-5℃~5℃,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,後處理得到白色固體中間體I。起始原料(苯、二苯硫醚或硫雜蒽酮)、AlCl3和R2基團的醯滷化合物的最佳摩爾比為1:1.1:1.05。
本發明中所述的有機溶劑A為二氯甲烷、二氯乙烷、氯仿或四氯化碳。
本發明中所述的步驟b中間體Ⅰ-B合成的具體操作為:有機溶劑B中加入中間體I,攪拌均勻後,室溫下加入鹽酸或通氯化氫,通亞硝酸甲酯或滴加亞硝酸異戊酯,室溫攪拌反應3~5h,減壓濃縮,重結晶得白色固體中間體Ⅱ。
本發明中所述的有機溶劑B為四氫呋喃、異丙醚或甲基叔丁基醚、乙醚、苯甲醚、丁醚、 乙二醇二乙醚及二氧六環。
本發明中所述的步驟c醯基肟酯光引發劑合成的具體操作為:向有機溶劑C中加入中間體Ⅰ-B和吡啶或三乙胺,攪拌均勻,冰鹽水浴冷卻至0℃左右開始滴加M1基團的醯滷化合物及有機溶劑C的混合液,1.5h左右滴加完畢,自然恢復至室溫繼續攪拌反應2h左右,後處理,得淺黃色油狀液體,即為本發明通式Ⅰ所示的醯基肟酯類化合物
本發明中所述的有機溶劑C為二氯甲烷、二氯乙烷、氯仿、四氯化碳或二氧六環。
本發明還提供了一種通式Ⅰ所述肟酯類化合物在UV光固化材料中的應用。
本發明的有益效果:本發明所述的肟酯類化合物在摩爾濃度相同的情況下,其紫外吸收效果與OXE-1的紫外吸收效果基本相近,其中本發明所述的肟酯類化合物的熱穩定性比OXE-1更加穩定;本發明所述的肟酯類化合物有部分的物質結構在紫外吸收圖譜中與OXE-1有明顯紅移,在300~365nm有較大吸收,可實現LED冷光源作為激活光源使用,本發明所述的肟酯類化合物的應用性能(感光度、熱穩定性、溶解性)比現有的OXE-1的應用性能好,同時,相比於現有同類產品,所述的肟酯類化合物在整體上表現出了顯著改善的綜合應用性能。
另外,本發明所述的肟酯類化合物還具有非常優異的儲存穩定性和很高的光固化活性,在低曝光劑量下,就能交聯固化且固化效果極佳,光固化過程不產生有毒、有害物質,使用安全性高。製得的膜邊緣平整無缺陷,沒有浮渣,整個圖案完整度好,表面沒有折皺,製成的彩色濾光片光學透明度高,不漏光。突出的表現在感光性組合物的氣味性、存儲穩定性、顯影性、成形膜的表面抗折皺性、使用安全性等方面,取得了非常好的技術效果。
附圖說明
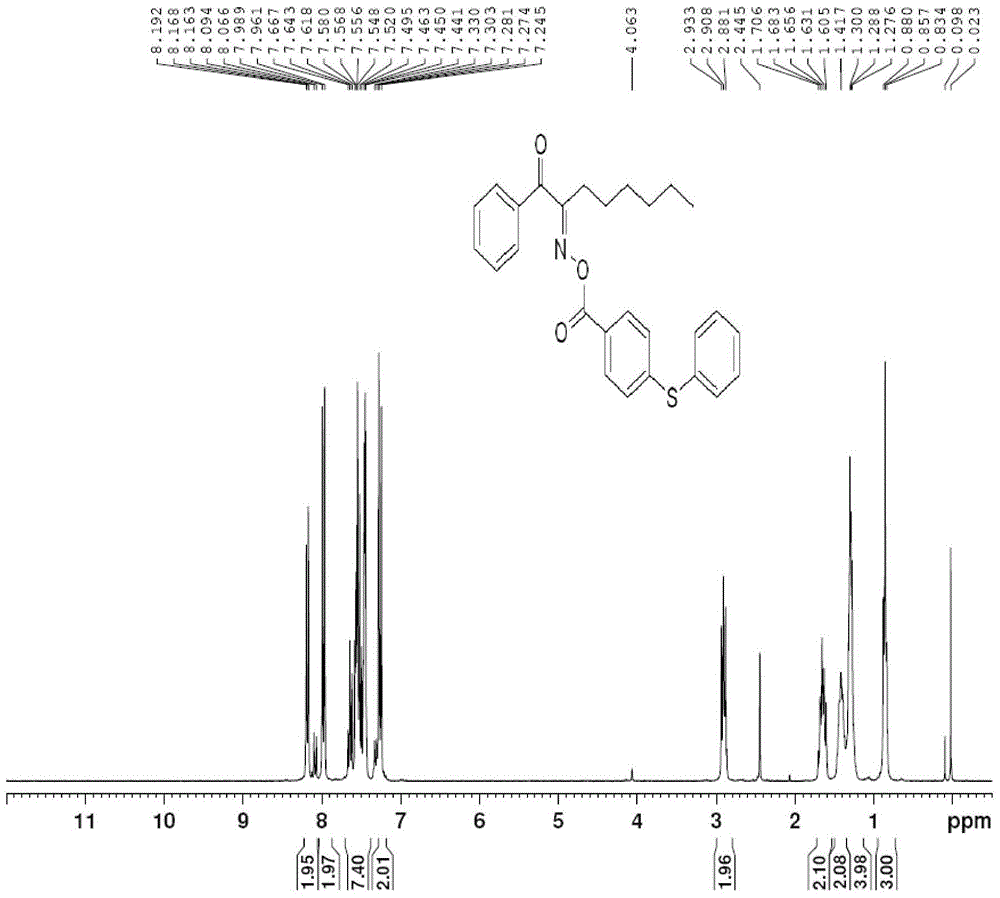
圖1為(E)-2-((羥亞氨基)-1-(苯基)辛-1-酮(化合物Ⅰ-1-1-3)核磁1HNMR譜圖。
圖2為(E)-1-苯基-2-((4-(苯巰基)苯甲醯氧基)亞氨基)辛-1-酮(化合物Ⅰ-1-1)的核磁1HNMR譜圖。
圖3為(E)-2-((羥亞氨基)-1-(4-(苯巰基)苯基)辛-1-酮(化合物Ⅰ-3-1-3)的核磁1HNMR譜圖。
圖4為(E)-2-((1,4-二(苯巰基)苯甲醯氧基)亞氨基)辛-1-酮(化合物Ⅰ-3-1)的核磁1HNMR譜圖。
圖5為(E)-2-((1-(苯巰基)苯甲醯氧基)亞氨基-4-(苯氧基)苯甲醯氧基)辛-1-酮(化合物Ⅰ-5-1)的核磁1HNMR譜圖。
圖6為1-氯-4-(2-羥亞胺基)辛酸酯基硫雜蒽酮(Ⅰ-8-1-3)的核磁1HNMR譜圖。
圖7為(E)-1-氯-(2-(3-苯巰基苯甲醯亞氨酯基))-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-8-1)的核磁1HNMR譜圖。
圖8為(E)-1-氯-(2-(3-苯氧基苯甲醯亞氨酯基))-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-10-1)的核磁1HNMR譜圖。
圖9為(E)-1-苯基-2-((4-(苯巰基)苯甲醯氧基)亞氨基)辛-1-酮(化合物Ⅰ-1-1)的UV譜圖。
圖10為(E)-2-((1,4-二(苯巰基)苯甲醯氧基)亞氨基)辛-1-酮(化合物Ⅰ-3-1)的UV譜圖。
圖11為為(E)-2-((1-(苯巰基)苯甲醯氧基)亞氨基-4-(苯氧基)苯甲醯氧基)辛-1-酮(化合物Ⅰ-5-1)的UV譜圖。
圖12為(E)-1-氯-(2-(3-苯巰基苯甲醯亞氨酯基))-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-8-1)的UV譜圖。
圖13為(E)-1-氯-(2-(3-苯氧基苯甲醯亞氨酯基))-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-10-1)的UV譜圖。
具體實施方式
本發明通式Ⅰ的化合物優選如下化合物中的一種或多種:
實施例1
正辛醯氯(Ⅰ-1-1-1)的製備
三口燒瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加165g氯化亞碸及30ml二氯甲烷混合液,約1h滴加完畢,回流2h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得淺黃色溶液114g,即為化合物Ⅰ-1-1-1。
1-(苯基)辛-1-酮(化合物Ⅰ-1-1-2)的製備
三口燒瓶,加入25.6g(0.33mol)苯和100ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入48.1g(0.34mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴56g(0.35mol)的正辛醯氯(Ⅰ-1-1-1)和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約1h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應液慢倒入10%的冰的稀鹽酸中,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓濃縮,然後過矽膠柱得黃色油狀液體57g,即為化合物Ⅰ-1-1-2,MS:m/z=204.2。
(E)-2-((羥亞氨基)-1-(苯基)辛-1-酮(化合物Ⅰ-1-1-3)的製備
三口燒瓶,加入5g(24.5mmol)化合物Ⅰ-1-1-2和30ml四氫呋喃溶液,室溫攪拌溶解,通入HCl氣體,室溫下滴加4.3g(36.8mmol)亞硝酸異戊酯,TLC監控至反應完全後,加入50ml二氯甲烷,2%NaHCO3溶液調至中性後分液,3×50ml水洗滌,無水MgSO4乾燥,過濾後減壓濃縮,得紅棕色油狀液體6g,即為化合物Ⅰ-1-1-3,MS:m/z=233.1。
1HNMR(300MHz,CDCl3),0.85~0.88(t,J=6.8Hz,3H),2.74~2.76(t,J=6.4Hz,2H),1.26-1.31(m,6H),1.54-1.56(m,2H),8.65(br,1H),7.41-7.45(d,J=9.2Hz,2H),7.52-7.56(m,1H),7.75-7.79(d,J=9.2Hz,2H),如圖1所示。
2-氯-1-(4-(苯巰基)苯基)乙酮(化合物Ⅰ-1-1-4)的製備
三口燒瓶,加入50g(0.27mol)二苯硫醚和200ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入39.4g(0.30mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴31.8g(0.28mol)的氯乙醯氯和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約1h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應液緩慢倒入10%的冰的稀鹽酸中,PH=4,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到45g黃色固體,即為化合物Ⅰ-1-1-4.
4-苯巰基苯甲酸(化合物Ⅰ-1-1-5)的製備
在三口瓶中,加入冷卻的25%NaOH溶液(1),NaClO溶液(10%)(1),三甲基苄基溴化銨,控溫0℃~5℃,控溫緩慢滴45g(0.17mol)的2-氯-1-(4-(苯巰基)苯基)已酮(化合物Ⅰ-1-4)和200ml二氯甲烷的混合液,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。加入亞硫酸氫鈉,攪拌30min後加入200ml二氯甲烷萃和10%的鹽酸,PH=3,分液,再用150ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌至中性後分液,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到30g黃色固體,即為化合物Ⅰ-1-1-5.
4-(苯巰基)苯甲醯氯(化合物Ⅰ-1-1-6)的製備
三口燒瓶,加入二氯甲烷100ml、4-(苯巰基)苯甲酸(Ⅰ-1-1-5)21g(0.091mol)以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加11.2g(0.095mol)氯化亞碸及30ml二氯甲烷混合液,約0.5h滴加完畢,回流1h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得棕黃色油狀液體26g,即為化合物Ⅰ-1-1-6。
(E)-1-苯基-2-((4-(苯巰基)苯甲醯氧基)亞氨基)辛-1-酮(化合物Ⅰ-1-1)的製備
加入23.3g(0.10mol)化合物Ⅰ-1-1-3和100ml二氯甲烷,降溫至0℃左右後加入11.7g(0.15mol)吡啶,控溫滴加27.2g(0.11mol)化合物Ⅰ-1-1-6及50ml二氯甲烷,滴加完畢後控溫0℃-5℃反應3h,反應完畢,加入100mlH20,加入飽和NaHCO3溶液洗至pH=5左右,3×100ml水洗至中性,無水MgSO4乾燥,過濾,減壓濃縮得橙黃色油狀液體42.5g,無水乙醇重結晶,得到29g淺黃色固體,收率為收率68%,即為化合物Ⅰ-1-1。
1HNMR(300MHz,CDCl3),0.83~0.85(t,J=6.9Hz,3H),1.27-1.30(t,J=7.2Hz,4H),1.41(s,J=6.2Hz,2H),1.60-1.70(m,2H),2.88-2.93(t,J=8.1Hz,2H),7.24-7.28(d,J=8.7Hz,2H),7.44-7.66(m,7H),7.96-7.98(m,2H),8.06~8.09(d,J=8.7Hz,2H),如圖2所示。
實施例2
正辛醯氯(Ⅰ-3-1-1)的製備
三口燒瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加165g氯化亞碸及30ml二氯甲烷混合液,約1h滴加完畢,回流2h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得淺黃色溶液114g,即為化合物Ⅰ-3-1-1。
1-(4-(苯巰基)苯基)辛-1-酮(化合物Ⅰ-3-1-2)的製備
三口燒瓶,加入123g(0.66mol)二苯硫醚和350ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入97.1g(0.73mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴113g(0.69mol)的正辛醯氯(Ⅰ-3-1-1)和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應物緩慢倒入200ml10%的冰的稀鹽酸中,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓濃縮,重結晶得白色固體175g,即為化合物Ⅰ-3-1-2,產率85%。MP:31-32℃;MS:m/z=312.15。
(E)-2-((羥亞氨基)-1-(4-(苯巰基)苯基)辛-1-酮(化合物Ⅰ-3-1-3)的製備
三口燒瓶,加入300ml氯化氫的四氫呋喃溶液(含29gHCl)和31.2g(0.1mol)化合物Ⅰ-3-1-2,室溫攪拌溶解,室溫下滴加150ml亞硝酸甲酯的四氫呋喃溶液(0.12mol),至反應完全後,減壓濃縮,得橙紅色油狀液體40g,加入250ml二氯甲烷,3×100ml水洗滌,無水MgSO4乾燥,過濾後減壓濃縮,得紅棕色油狀液體34g,加入120ml石油醚,攪拌下加熱溶解完全, 緩慢降溫至-5℃,析出白色固體,抽濾,石油醚洗濾餅,得白色固體18.7g,即為化合物Ⅰ-3-1-3,產率55%。
1HNMR(300MHz,CDCl3),0.88(t,J=6.6Hz,3H),2.73(t,J=7.5Hz,2H),1.29-1.36(m,6H),1.50-1.58(m,2H),8.73(br,1H),7.18-7.28(d,J=4.2Hz,2H),7.39-7.44(m,3H),7.50-7.53(m,2H),7.78-7.87(d,J=8.7Hz,2H),如圖3所示。
2-氯-1-(4-(苯巰基)苯基)乙酮(化合物Ⅰ-3-1-4)的製備
三口燒瓶,加入50g(0.27mol)二苯硫醚和200ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入39.4g(0.30mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴31.8g(0.28mol)的氯乙醯氯和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約1h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應液緩慢倒入10%的冰的稀鹽酸中,PH=4,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到45g黃色固體,即為化合物Ⅰ-3-1-4.
4-苯巰基苯甲酸(化合物Ⅰ-3-1-5)的製備
在三口瓶中,加入冷卻的25%NaOH溶液,NaClO溶液(10%),三甲基苄基溴化銨,控溫0℃~5℃,控溫緩慢滴45g(0.17mol)的2-氯-1-(4-(苯巰基)苯基)已酮(化合物Ⅰ-3-1-4)的和200ml二氯甲烷的混合液,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。加入亞硫酸氫鈉,攪拌30min後加入200ml二氯甲烷萃和10%的鹽酸,PH=3,分液,再用150ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌至中性後分液,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到30g黃色固體,即為化合物Ⅰ-3-1-5.
4-(苯巰基)苯甲醯氯(化合物Ⅰ-3-1-6)的製備
三口燒瓶,加入二氯甲烷100ml、4-(苯巰基)苯甲酸(Ⅰ-3-1-5)21g(0.091mol)以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加11.2g(0.095mol)氯化亞碸及30ml二氯甲烷混合液,約0.5h滴加完畢,回流1h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得棕黃色油狀液體26g,即為化合物Ⅰ-3-1-6。
(E)-2-((1,4-二(苯巰基)苯甲醯氧基)亞氨基)辛-1-酮(化合物Ⅰ-3-1)的製備
加入34.1g(0.10mol)化合物Ⅰ-3-1-3和100ml二氯甲烷,降溫至0℃左右後加入11.7g(0.15mol)吡啶,控溫滴加27.2g(0.11mol)化合物Ⅰ-3-1-6及50ml二氯甲烷,滴加完畢後控溫0℃-5℃反應3h,反應完畢,加入100mlH20,,加入飽和NaHCO3溶液洗至pH=5左右,3×100ml水洗至中性,無水MgSO4乾燥,過濾,減壓濃縮得橙黃色油狀液體50g,無水乙醇重結晶,得到41g淺黃色固體,收率為收率75%,即為化合物Ⅰ-3-1。
1HNMR(300MHz,CDCl3),0.84~0.86(t,J=6.9Hz,3H),1.25-1.28(t,J=7.2Hz,4H),1.38(s,J=6.2Hz,2H),1.59-1.64(m,2H),2.84-2.89(t,J=8.1Hz,2H),7.20-7.28(d,J=8.7Hz,2H),7.42-7.45(m,5H),7.53-7.56(m,4H),7.93~7.96(d,J=8.7Hz,2H),8.03~8.06(d,J=8.5Hz,2H),如圖4所示。
實施例3
正辛醯氯(Ⅰ-5-1-1)的製備
三口燒瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加165g氯化亞碸及30ml二氯甲烷混合液,約1h滴加完畢,回流2h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得淺黃色溶液114g,即為化合物Ⅰ-5-1-1。
1-(4-(苯巰基)苯基)辛-1-酮(化合物Ⅰ-5-1-2)的製備
三口燒瓶,加入123g(0.66mol)二苯硫醚和350ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入97.1g(0.73mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴113g(0.69mol)的正辛醯氯(Ⅰ-5-1-1)和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應物緩慢倒入200ml10%的冰的稀鹽酸中,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓濃縮,重結晶得白色固體175g,即為化合物Ⅰ-5-1-2,產率85%。MP:31-32℃; MS:m/z=312.15。
(E)-2-((羥亞氨基)-1-(4-(苯巰基)苯基)辛-1-酮(化合物Ⅰ-5-1-3)的製備
三口燒瓶,加入300ml氯化氫的四氫呋喃溶液(含29gHCl)和31.2g(0.1mol)化合物Ⅰ-3-1-2,室溫攪拌溶解,室溫下滴加150ml亞硝酸甲酯的四氫呋喃溶液(0.12mol),至反應完全後,減壓濃縮,得橙紅色油狀液體40g,加入250ml二氯甲烷,3×100ml水洗滌,無水MgSO4乾燥,過濾後減壓濃縮,得紅棕色油狀液體34g,加入120ml石油醚,攪拌下加熱溶解完全,緩慢降溫至-5℃,析出白色固體,抽濾,石油醚洗濾餅,得白色固體18.7g,即為化合物Ⅰ-5-1-3,產率55%。
1HNMR(300MHz,CDCl3),0.88(t,J=6.6Hz,3H),2.73(t,J=7.5Hz,2H),1.29-1.36(m,6H),1.50-1.58(m,2H),8.73(br,1H),7.18-7.28(d,J=4.2Hz,2H),7.39-7.44(m,3H),7.50-7.53(m,2H),7.78-7.87(d,J=8.7Hz,2H)。
2-氯-1-(4-(苯氧基)苯基)乙酮(化合物Ⅰ-5-1-4)的製備
三口燒瓶,加入50g(0.29mol)二苯醚和200ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入42.1g(0.32mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴34.1g(0.30mol)的氯乙醯氯和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約1h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應液緩慢倒入10%的冰的稀鹽酸中,PH=4,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到48g黃色固體,即為化合物Ⅰ-5-1-4.
4-苯氧基苯甲酸(化合物Ⅰ-5-1-5)的製備
在三口瓶中,加入冷卻的25%NaOH溶液,NaClO溶液(10%),三甲基苄基溴化銨,控溫0℃~5℃,控溫緩慢滴45g(0.18mol)的Ⅰ-5-1-4和200ml二氯甲烷的混合液,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。加入亞硫酸氫鈉,攪拌30min後加入200ml二氯甲烷萃和10%的鹽酸,PH=3,分液,再用150ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌至中性後分液,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到29g黃色固體,即為化合物Ⅰ-5-1-5.
4-(苯氧基)苯甲醯氯(化合物Ⅰ-5-1-6)的製備
三口燒瓶,加入二氯甲烷100ml、4-苯氧基苯甲酸(Ⅰ-5-1-5)20g(0.093mol)以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加16.7g(0.14mol)氯化亞碸及30ml二氯甲烷混合液,約0.5h滴加完畢,回流1h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得黃色油狀液體23g,即為化合物Ⅰ-5-1-6。
(E)-2-((1-(苯巰基)苯甲醯氧基)亞氨基-4-(苯氧基)苯甲醯氧基)辛-1-酮(化合物Ⅰ-5-1)的製備
加入34.1g(0.10mol)化合物Ⅰ-5-1-3和100ml二氯甲烷,降溫至0℃左右後加入11.7g(0.15mol)吡啶,控溫滴加25.5g(0.11mol)化合物Ⅰ-5-1-6及50ml二氯甲烷,滴加完畢後控溫0℃-5℃反應3h,反應完畢,加入100mlH20,,加入飽和NaHCO3溶液洗至pH=5左右,3×100ml水洗至中性,無水MgSO4乾燥,過濾,減壓濃縮得橙黃色油狀液體48g,無水乙醇重結晶,得到39g淺黃色固體,收率為收率73%,即為化合物Ⅰ-5-1。
1HNMR(300MHz,CDCl3),0.83~0.85(t,J=6.9Hz,3H),1.24-1.27(t,J=7.2Hz,4H),1.38(s,J=6.2Hz,2H),1.56-1.64(m,2H),2.84-2.89(t,J=8.1Hz,2H),7.02-7.08(m,4H),7.19-7.25(m,3H),7.39-7.41(m,5H),7.51~7.53(d,J=5.7Hz,2H),7.91~7.94(s,J=8.5Hz,1H),8.03~8.06(m,3H),如圖5所示。
實施例4
正辛醯氯(化合物Ⅰ-8-1-1)的製備
三口燒瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加165g氯化亞碸及30ml二氯甲烷混合液,約1h滴加完畢,回流2h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得淺黃色溶液114g,即為化合物Ⅰ-8-1-1。
1-氯-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-8-1-2)的製備
加入5.24g(0.02mol)化合物1-氯-4-羥基硫雜蒽酮和50ml二氯甲烷,降溫至0℃左右後加入2.37g(0.03mol)吡啶,控溫滴加3.56g(0.022mol)化合物Ⅰ-8-1-1及20ml二氯甲烷,滴加完畢後自然升溫反應3h,TLC板跟蹤至反應完畢,加入30mlH20,攪拌30min後分液,加入飽和NaHCO3溶液100ml洗至pH=10左右,水洗滌至中性,再加入2%稀鹽酸調至pH=4左右,3×100ml水洗至中性,無水MgSO4乾燥,過濾,減壓濃縮得棕黃色固體,乙醇重結晶得6.5g,收率84%,即為化合物Ⅰ-8-1-2。
1-氯-4-(2-羥亞胺基)辛酸酯基硫雜蒽酮(Ⅰ-8-1-3)的製備
三口燒瓶,加入300ml氯化氫的四氫呋喃溶液(含29gHCl)和38.9g(0.1mol)化合物Ⅰ-8-1-2,室溫攪拌溶解,室溫下滴加150ml亞硝酸甲酯的四氫呋喃溶液(0.12mol),至反應完全後,減壓濃縮,得橙紅色油狀液體49g,加入250ml二氯甲烷,3×100ml水洗滌,無水MgSO4乾燥,過濾後減壓濃縮,得紅棕色油狀液體41g,加入120ml石油醚,攪拌下加熱溶解完全,緩慢降溫至-5℃,析出土黃色固體,抽濾,石油醚洗濾餅,得土黃色固體26.7g,即為化合物Ⅰ-8-1-3,產率64%。
1HNMR(300MHz,CDCl3),0.85~0.88(t,J=7.3Hz,3H),2.71~2.75(t,J=9.4Hz,2H),1.30-1.36(m,6H),1.50-1.57(m,2H),11.24(br,1H),7.21(s,J=7.2Hz,1H),7.26~7.31(s,J=7.2Hz,1H),7.46(s,J=8.1Hz,1H),7.54-7.57(d,J=8.9Hz,2H),8.24-8.26(s,J=7.2Hz,1H),如圖6所示。
2-氯-1-(4-(苯巰基)苯基)乙酮(化合物Ⅰ-8-1-4)的製備
三口燒瓶,加入50g(0.27mol)二苯硫醚和200ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入39.4g(0.30mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴31.8g(0.28mol)的氯乙醯氯和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約1h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應液緩慢倒入10%的冰的稀鹽酸中,PH=4,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓 濃縮,重結晶後得到45g黃色固體,即為化合物Ⅰ-8-1-4.
4-苯巰基苯甲酸(化合物Ⅰ-8-1-5)的製備
在三口瓶中,加入冷卻的25%NaOH溶液,NaClO溶液(10%),三甲基苄基溴化銨,控溫0℃~5℃,控溫緩慢滴45g(0.17mol)的Ⅰ-8-1-4和200ml二氯甲烷的混合液,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。加入亞硫酸氫鈉,攪拌30min後加入200ml二氯甲烷萃和10%的鹽酸,PH=3,分液,再用150ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌至中性後分液,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到30g黃色固體,即為化合物Ⅰ-8-1-5.
4-(苯巰基)苯甲醯氯(化合物Ⅰ-8-1-6)的製備
三口燒瓶,加入二氯甲烷100ml、4-(苯巰基)苯甲酸21g(0.091mol)以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加11.2g(0.095mol)氯化亞碸及30ml二氯甲烷混合液,約0.5h滴加完畢,回流1h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得棕黃色油狀液體26g,即為化合物Ⅰ-8-1-6。
(E)-1-氯-(2-(3-苯巰基苯甲醯亞氨酯基))-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-8-1)的製備
加入8.36g(0.02mol)化合物Ⅰ-8-1-3和50ml二氯甲烷,降溫至0℃左右後加入2.37g(0.03mol)吡啶,控溫滴加5.45g(0.022mol)化合物Ⅰ-8-1-6及20ml二氯甲烷,滴加完畢後自然升溫反應3h,反應完畢,加入30mlH20,攪拌30min後分液,加入飽和NaHCO3溶液100ml洗至pH=10左右,水洗滌至中性,再加入2%稀鹽酸調至pH=4左右,3×100ml水洗至中性,無水MgSO4乾燥,過濾,減壓濃縮得土黃色固體,乙醇重結晶得10g,收率80%,即為化合物Ⅰ-8-1。
1HNMR(300MHz,CDCl3),0.85~0.88(t,J=6.4Hz,3H),2.83~2.85(t,J=8.5Hz,2H),1.30-1.33(m,4H),1.43-1.47(m,2H),1.63~1.68(m,2H),7.36~7.38(d,J=7.4Hz,2H),7.44~7.50(m,6H),7.54~7.58(m,4H),8.12~8.15(d,J=9.2Hz,2H),8.43~8.45(s,J=8.4Hz, 1H),如圖7所示。
實施例5
正辛醯氯(化合物Ⅰ-10-1-1)的製備
三口燒瓶,加入二氯甲烷200ml、正辛酸100g以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加165g氯化亞碸及30ml二氯甲烷混合液,約1h滴加完畢,回流2h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得淺黃色溶液114g,即為化合物Ⅰ-10-1-1。
1-氯-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-10-1-2)的製備
加入5.24g(0.02mol)化合物1-氯-4-羥基硫雜蒽酮和50ml二氯甲烷,降溫至0℃左右後加入2.37g(0.03mol)吡啶,控溫滴加3.56g(0.022mol)化合物Ⅰ-10-1-1及20ml二氯甲烷,滴加完畢後自然升溫反應3h,TLC板跟蹤至反應完畢,加入30mlH20,攪拌30min後分液,加入飽和NaHCO3溶液100ml洗至pH=10左右,水洗滌至中性,再加入2%稀鹽酸調至pH=4左右,3×100ml水洗至中性,無水MgSO4乾燥,過濾,減壓濃縮得棕黃色固體,乙醇重結晶得6.5g,收率84%,即為化合物Ⅰ-10-1-2。
1-氯-4-(2-羥亞胺基)辛酸酯基硫雜蒽酮(Ⅰ-10-1-3)的製備
三口燒瓶,加入300ml氯化氫的四氫呋喃溶液(含29gHCl)和38.9g(0.1mol)化合物Ⅰ-10-1-2,室溫攪拌溶解,室溫下滴加150ml亞硝酸甲酯的四氫呋喃溶液(0.12mol),至反應完全後,減壓濃縮,得橙紅色油狀液體49g,加入250ml二氯甲烷,3×100ml水洗滌,無水MgSO4乾燥,過濾後減壓濃縮,得紅棕色油狀液體41g,加入120ml石油醚,攪拌下加熱溶解完全,緩慢降溫至-5℃,析出棕土黃色固體,抽濾,石油醚洗濾餅,得土黃色固體26.7g,即為化合物Ⅰ-10-1-3,產率64%。
1HNMR(300MHz,CDCl3),0.85~0.88(t,J=7.3Hz,3H),2.71~2.75(t,J=9.4Hz,2H),1.30-1.36(m,6H),1.50-1.57(m,2H),11.24(br,1H),7.21(s,J=7.2Hz,1H),7.26~7.31(s,J=7.2Hz,1H),7.46(s,J=8.1Hz,1H),7.54-7.57(d,J=8.9Hz,2H),8.24-8.26(s,J=7.2 Hz,1H)。
2-氯-1-(4-(苯氧基)苯基)乙酮(化合物Ⅰ-10-1-4)的製備
三口燒瓶,加入50g(0.29mol)二苯醚和200ml二氯甲烷,冰鹽水浴降溫至-5℃,通氮氣,加入42.1g(0.32mol)無水A1C13,加乾燥管、回流冷凝管及尾氣吸收,緩慢滴34.1g(0.30mol)的氯乙醯氯和50ml二氯甲烷的混合液,控制溫度在-5~5℃,約1h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。將反應液緩慢倒入10%的冰的稀鹽酸中,PH=4,攪拌30min後分液,100ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌,2%NaHCO3溶液調至中性後分液,100ml水洗1次,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到48g黃色固體,即為化合物Ⅰ-10-1-4.
4-苯氧基苯甲酸(化合物Ⅰ-10-1-5)的製備
在三口瓶中,加入冷卻的25%NaOH溶液,NaClO溶液(10%),三甲基苄基溴化銨,控溫0℃~5℃,控溫緩慢滴45g(0.18mol)的Ⅰ-10-1-4和200ml二氯甲烷的混合液,約2h滴加完畢,撤去冰鹽水浴,自然恢復至室溫,繼續攪拌反應2~3h,TLC監控反應完全。加入亞硫酸氫鈉,攪拌30min後加入200ml二氯甲烷萃和10%的鹽酸,PH=3,分液,再用150ml二氯甲烷萃取水相,合併有機相,3×100ml水洗滌至中性後分液,無水MgSO4乾燥,過濾,減壓濃縮,重結晶後得到29g黃色固體,即為化合物Ⅰ-10-1-5.
4-(苯氧基)苯甲醯氯(化合物Ⅰ-10-1-6)的製備
三口燒瓶,加入二氯甲烷100ml、4-苯氧基苯甲酸(Ⅰ-10-1-5)20g(0.093mol)以及2滴DMF,搭上回流冷凝管、恆壓滴液漏鬥、乾燥管及鹼液尾氣吸收裝置,加熱至回流,緩慢滴加16.7g(0.14mol)氯化亞碸及30ml二氯甲烷混合液,約0.5h滴加完畢,回流1h,減壓濃縮,加入新鮮二氯甲烷100ml再次減壓濃縮,得黃色油狀液體23g,即為化合物Ⅰ-10-1-6。
(E)-1-氯-(2-(3-苯氧基苯甲醯亞氨酯基))-4-辛酸酯基硫雜蒽酮(化合物Ⅰ-10-1)的製備
加入8.36g(0.02mol)化合物Ⅰ-10-1-3和50ml二氯甲烷,降溫至0℃左右後加入2.37g(0.03mol)吡啶,控溫滴加5.1g(0.022mol)化合物Ⅰ-10-1-6及20ml二氯甲烷,滴加完畢後自然升溫反應3h,反應完畢,加入30mlH20,攪拌30min後分液,加入飽和NaHCO3溶液100ml洗至pH=10左右,水洗滌至中性,再加入2%稀鹽酸調至pH=4左右,3×100ml水洗至中性,無水MgSO4乾燥,過濾,減壓濃縮得土黃色固體,乙醇重結晶得10g,收率80%,即為化合物Ⅰ-10-1。
1HNMR(300MHz,CDCl3),0.86~0.88(t,J=6.5Hz,3H),2.65~2.70(t,J=8.5Hz,2H),1.30~1.40(m,6H),1.49~1.57(m,2H),7.13~7.22(m,4H),7.36(s,J=7.4Hz,1H),7.42~7.55(m,5H),7.58~7.61(m,2H),8.25~8.28(d,J=7.9Hz,2H),8.43~8.46(s,J=8.4Hz,1H),如圖8所示。
應用實施例1
檢測了化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1在常用的有機溶劑,如乙酸乙酯、乙二醇單乙醚、丙二醇甲醚、丙二醇甲醚醋酸酯中的溶解性。分別取五份等量的化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1置於五個試樣瓶中,分別用乙二醇單乙醚、丙二醇甲醚醋酸酯溶解,通過TLC檢測光引發劑在溶液中的穩定性。結果顯示均具有一定的溶解性,且此類光引發劑在有機溶劑中的穩定性比較好,避光條件下在常用溶劑中均能保持20天以上不發生分解。
應用實施例2
利用差熱-熱重分析儀測定化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1的熱解性能。升溫速度:10℃/min。檢測了化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1初始分解溫度均在140℃以上,熱穩定性較好。
應用實施例3
將化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1溶解在乙腈中,分別配成一定摩爾濃度的溶液,通過用紫外-可見分光光度計測其紫外吸收譜圖,進行比較。通過檢測譜圖可以看出所合成的幾種化合物紫外吸收譜帶較寬泛,在300~365nm均有較大吸收,如圖9、圖10、圖11、圖12及圖13所示。
應用實施例4
利用實時紅外測試手段,比較了實施例中化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1引發甲基丙烯酸羥乙酯聚合的雙鍵轉化率。以丙酮為溶劑,配製化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、 Ⅰ-8-1及Ⅰ-10-1的濃度為單體濃度的3%的試樣,塗於KBr鹽片上,然後放入Nico-let5700中,用紫外光點光源照射樣品,調節樣品表面的紫外光光強為30mW/cm2。單體的雙鍵轉化率用近紅外實時採集,實時紅外參數設置為數據採集間隔0.3985s,每個光譜掃描1次,解析度為4cm-1。甲基丙烯酸羥乙酯在近紅外譜圖中碳碳雙鍵的特徵吸收峰在1630cm-1處,隨著光固化反應的進行,碳碳雙鍵變成碳碳單鍵,雙鍵的吸收峰強度隨光照時間增加而變弱,所以利用碳碳雙鍵的特徵吸收峰的變化來反映其聚合反應的變化程度。雙鍵轉化率(DC)由數據處理軟體結合下式計算得到。
DC(%)=[1-(At/Ao)]×100%
式中Ao和At分別為樣品在固化前和光照後t時刻在1630cm-1處甲基丙烯酸羥乙酯雙鍵特徵吸收峰的面積。檢測結果顯示化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1均可以引發甲基丙烯酸羥乙酯聚合,在光照l0min之後,均能使丙烯酸雙鍵的轉化率達60%以上。
應用實施例5
用聚乙烯吡咯烷酮(MW=40000)與聚甲基丙烯酸酯樹脂(Mn=50000)作成膜樹脂,2-羥丁基丙烯酸酯作聚合單體,分別加入化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1,加入適量的鏈轉移劑和染料,用丙二醇甲醚作溶劑,配成自由基聚合成像感光膠液1、2、3、4、5。將以上膠液按以下方法進行性能測試。將感光膠液1~5用離心機旋塗在預先處理好的並滿足下列條件的PS鋁版基上,鋁板基尺寸:1030mm×800mm;鋁板基厚度:0.28~0.3mm;砂目規格:Ra=0.5~0.6μm,Rh=0.3~0.35μm;陽極氧化膜重量:3~3.5g/m2。控制離心塗布機的轉速,使塗在鋁版基上的塗布量(以固含量計)為0.5~2.5g/m2,在離心塗布機上初步乾燥後,轉移到100℃的鼓風乾燥機中乾燥3min,得紫雷射CTP原版。將原版經紫雷射曝光,用Ugra測試條做掩膜測試版材的感光性能。曝光後,用1%NaOH水溶液顯影。曝光區,可光聚合化合物在引發劑存在下發生聚合反應,顯影液中不溶,而非曝光區是可溶的,於是得到陰圖。通過曝光顯影,從得到的圖像的的連續調梯尺評價感度,從微線條測試塊區域評價精度,從而評價感光組合物感光性能的優劣。實驗結果顯示在曝光時間均為40s的情況下,化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1的版材均能顯到連續調梯尺的3段以上,精度在6μ以上。由此可見,化合物Ⅰ-1-1、Ⅰ-3-1、Ⅰ-5-1、Ⅰ-8-1及Ⅰ-10-1在一定曝光時間及曝光量下,能達到較好的成像效果,可很好的適用於紫雷射成像體系中。