一種多取代吡啶的製備方法與流程
2023-10-27 15:10:52 3
本發明涉及一種多取代吡啶的製備方法,屬於精細化工產品的製備領域。
背景技術:
吡啶是一類重要的雜環化合物,不但廣泛分布於自然界中,而且在藥物化學,合成化學,以及材料化學中都有廣泛的應用。例如,含吡啶結構的維生素B3(煙酸)和維生素B6(吡哆素)是人和動物內所必須的維生素,是多種輔酶的重要組成部分,用於參與體內的重要代謝活動。NADP也是生物體內一類重要的含吡啶結構的輔酶,在呼吸作用中參與氧化還原過程提供能量。除此之外,在已上市的暢銷藥物(如吡格列酮,雷貝拉唑,伊馬替尼等),pH值螢光指示材料【J.Am.Chem.Soc.2009,131,3016】,以及手型多齒配體【Org.Lett.2007,9,3933】中,均可發現多取代吡啶的結構。因此,由於其在自然界和工業中的重要性,人們希望找到更多綠色環保、合成簡便的方法製備該類雜環化合物。
合成吡啶最常用的方法是使用胺與1,5-二羰基化合物縮合,再通過氧化作用將縮合產物二氫吡啶氧化形成多取代吡啶。而通常使用的氧化劑為硝酸【Liebigs Ann.Chem.1882,215,1】,1,4-二苯醌【Eur.J.Org.Chem.2001,2115】,或者單質碘【Synthesis,2000,1532.】,當然使用氧氣或者空氣作為氧化劑也有相關文獻報導,然而通常都需要200度以上的高溫【J.Org.Chem.2002,2197】。由於這些氧化過程需要消耗大量的氧化劑以及高溫反應不易操作的缺陷,因此能夠尋求一些廉價易得、操作簡便、綠色環保的氧化劑完成1,5-二羰基化合物到吡啶的轉化十分必要。
二甲基亞碸(DMSO)既是一種常見的有機溶劑,又是一類綠色環保的氧化劑。例如經典的氧化反應Swern氧化,就是利用二甲基亞碸的氧化性將伯醇氧化成醛。
技術實現要素:
本發明旨在提供一種多取代吡啶的製備方法,反應條件溫和,操作過程簡單。
本發明提供了一種多取代吡啶的製備方法,使用1,5-二羰基化合物和醋酸銨為原料,不需要外加任何氧化劑,以二甲基亞碸為溶劑和氧化劑,加熱至85-95℃得到1,2,3,5-四取代的吡啶產品;收率可達70-87%;所使用的1,5-二羰基化合物結構如下:
其中R1和R2代表具有不同取代基的苯環;R3代表酯基;R4代表甲基或有不同取代基的苯環。
本發明關於取代基和苯環的定義均為本領域普通技術人員所熟知,在此及下文不再做詳細的說明。
本發明的反應方程式如下:
上述多取代吡啶的製備方法,具體操作步驟如下:
(1)將1,5-二羰基化合物溶解在DMSO中,攪拌使其溶解,稱取醋酸銨固體一次性加入上述DMSO溶液中,將混合液升溫至85-95℃,攪拌8-12小時;
(2)通過薄層層析板檢查反應直至原料1,5-二羰基化合物反應完全,反應混合液降至室溫用乙酸乙酯稀釋,並用去離子水洗滌乙酸乙酯層3~5次;乙酸乙酯層有機相減壓蒸乾,濃縮物用二氯甲烷重結晶,製得1,2,3,5-四取代的吡啶產品。
上述製備方法中,底物1,5-二羰基化合物與醋酸銨的摩爾比為1:(3-10);每摩爾1,5-二羰基化合物所使用的溶劑DMSO的體積為5-10升。
上述原料配比優選為底物1,5-二羰基化合物與醋酸銨的摩爾比為1:5;每摩爾1,5-二羰基化合物使用溶劑DMSO的體積為5升。
上述製備方法中,反應最佳條件為:反應溫度為90℃,反應時間為8小時。
本發明為1,2,3,5-四取代吡啶特別是3位具有酯基的取代吡啶提供了一種簡便、綠色、高效的合成方法。
本發明的反應機理如下所述:醋酸銨在加熱的條件下釋放出氨氣分子與含有酯基的1,5-二羰基化合物縮合形成烯胺,烯胺發生分子內縮合併通過異構化形成二氫吡啶;在DMSO的氧化作用下,二氫吡啶被氧化,並最終脫去水分子形成吡啶。
本發明的有益效果:
本方法使用二甲基亞碸(DMSO)為溶劑和氧化劑,在加熱的條件下可以完成1,5-二羰基化合物與醋酸銨反應生成1,2,3,5-四取代吡啶。反應體系簡單,避免了額外添加氧化劑以及高溫強熱的條件,尤其針對於3位有酯基取代的吡啶結構有較好的適用性。
附圖說明
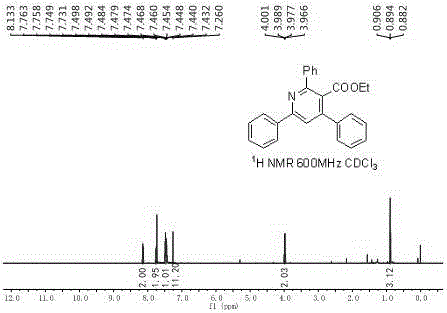
圖1為實施例1產品的核磁共振氫譜圖。
圖2為實施例2產品的核磁共振氫譜圖。
圖3為實施例3產品的核磁共振氫譜圖。
圖4為實施例4產品的核磁共振氫譜圖。
圖5為實施例5產品的核磁共振氫譜圖。
圖6為實施例6產品的核磁共振氫譜圖。
圖7為實施例7產品的核磁共振氫譜圖。
具體實施方式
下面通過實施例來進一步說明本發明,但不局限於以下實施例。
實施例1:
在500mL的圓底燒瓶中,加入20g(50mmol)2-苯甲醯基-5-氧雜-3,5-二苯基戊酸乙酯,攪拌使其溶解於250mL二甲基亞碸中。將20g(250mmol)醋酸銨固體加入到上述混合液中,室溫攪拌使其溶解。待醋酸銨完全溶解後,將反應體系升至90℃攪拌8小時,並用薄層層析板檢測反應進度。
反應結束,停止攪拌並將反應液降至室溫,加入500mL乙酸乙酯稀釋反應液,並加入蒸餾水洗滌乙酸乙酯層3-5次,每次用水250mL。洗滌結束加入乾燥劑乾燥有機相,濃縮後用二氯甲烷重結晶得到白色固體15.2g,產率80%,熔點86-88℃。
所得產物經核磁鑑定為目標產品。
如圖1所示,1H NMR(CDCl3,600MHz):δ8.15-8.12(m,2H),7.77-7.74(m,2H),7.73(s,1H),7.51-7.43(m,11H),3.98(q,2H,J=7.2Hz),0.89(t,3H,J=7.2Hz)。
實施例2
在500mL的圓底燒瓶中,加入21g(50mmol)2-苯甲醯基-5-氧雜-3-苯基-5-對甲苯基戊酸乙酯,攪拌使其溶解於350mL二甲基亞碸中。將12g(150mmol)醋酸銨固體加入到上述混合液中,室溫攪拌使其溶解。待醋酸銨完全溶解後,將反應體系升至85℃攪拌10小時,並用薄層層析板檢測反應進度。
反應結束,停止攪拌並將反應液降至室溫,加入500mL乙酸乙酯稀釋反應液,並加入蒸餾水洗滌乙酸乙酯層3-5次,每次用水250mL。洗滌結束加入乾燥劑乾燥有機相,濃縮後用二氯甲烷重結晶得到白色固體14.3g,產率73%,熔點77-79℃。
所得產物經核磁鑑定為目標產品。
如圖2所示,1H NMR(CDCl3,400MHz):δ8.05(d,2H,J=8.0Hz),7.75(d,2H,J=6.4Hz),7.70(s,1H),7.52-7.41(m,8H),7.30(d,2H,J=7.6Hz),3.98(q,2H,J=7.2Hz),2.42(s,3H,),0.89(q,3H,J=7.2Hz)。
實施例3
在1L的圓底燒瓶中,加入22g(50mmol)2-苯甲醯基-5-氧雜-3-苯基-5-對硝基苯基戊酸乙酯,攪拌使其溶解於500mL二甲基亞碸中。將39g(500mmol)醋酸銨固體加入到上述混合液中,室溫攪拌使其溶解。待醋酸銨完全溶解後,將反應體系升至95℃攪拌12小時,並用薄層層析板檢測反應進度。
反應結束,停止攪拌並將反應液降至室溫,加入1L乙酸乙酯稀釋反應液,並加入蒸餾水洗滌乙酸乙酯層3-5次,每次用水250mL。洗滌結束加入乾燥劑乾燥有機相,濃縮後用二氯甲烷重結晶得到白色固體15.9g,產率75%,熔點151-153℃。
所得產物經核磁鑑定為目標產品。
如圖3所示,1H NMR(CDCl3,400MHz):δ8.36-8.30(m,4H),7.80(s,1H),7.76-7.72(m,2H),7.51-7.45(m,8H),4.00(q,2H,J=7.2Hz),0.90(t,3H,J=7.2Hz)。
實施例4
在500mL的圓底燒瓶中,加入21.5g(50mmol)2-苯甲醯基-5-氧雜-3-對甲氧基苯基-5-苯基戊酸乙酯,攪拌使其溶解於400mL二甲基亞碸中。將31g(400mmol)醋酸銨固體加入到上述混合液中,室溫攪拌使其溶解。待醋酸銨完全溶解後,將反應體系升至85℃攪拌10小時,並用薄層層析板檢測反應進度。
反應結束,停止攪拌並將反應液降至室溫,加入500mL乙酸乙酯稀釋反應液,並加入蒸餾水洗滌乙酸乙酯層3-5次,每次用水250mL。洗滌結束加入乾燥劑乾燥有機相,濃縮後用二氯甲烷重結晶得到白色固體14.3g,產率70%,熔點148-150℃。
所得產物經核磁鑑定為目標產品。
如圖4所示,1H NMR(CDCl3,400MHz):δ8.04(d,2H,J=8.0Hz),7.77-7.72(m,2H),7.70(s,1H),7.50-7.41(m,8H),7.29(d,2H,J=8.0Hz),3.98(q,2H,J=7.2Hz),2.42(s,3H),0.89(t,3H,J=7.2Hz)。
實施例5
在500mL的圓底燒瓶中,加入21.7g(50mmol)2-苯甲醯基-5-氧雜-3-對氯苯基-5-苯基戊酸乙酯,攪拌使其溶解於300mL二甲基亞碸中。將20g(250mmol)醋酸銨固體加入到上述混合液中,室溫攪拌使其溶解。待醋酸銨完全溶解後,將反應體系升至90℃攪拌10小時,並用薄層層析板檢測反應進度。
反應結束,停止攪拌並將反應液降至室溫,加入500mL乙酸乙酯稀釋反應液,並加入蒸餾水洗滌乙酸乙酯層3-5次,每次用水250mL。洗滌結束加入乾燥劑乾燥有機相,濃縮後用二氯甲烷重結晶得到白色固體18.0g,產率87%,熔點122-124℃。
所得產物經核磁鑑定為目標產品。
如圖5所示,1H NMR(CDCl3,600MHz):δ8.14-8.10(m,2H),7.76-7.72(m,2H),7.68(s,1H),7.51-7.40(m,10H),3.99(q,2H,J=7.2Hz),0.92(t,3H,J=7.2Hz)。
實施例6
在500mL的圓底燒瓶中,加入16.9g(50mmol)2-乙醯基-5-氧雜-3,5-二苯基戊酸乙酯,攪拌使其溶解於250mL二甲基亞碸中。將16g(200mmol)醋酸銨固體加入到上述混合液中,室溫攪拌使其溶解。待醋酸銨完全溶解後,將反應體系升至90℃攪拌8小時,並用薄層層析板檢測反應進度。
反應結束,停止攪拌並將反應液降至室溫,加入500mL乙酸乙酯稀釋反應液,並加入蒸餾水洗滌乙酸乙酯層3-5次,每次用水250mL。洗滌結束加入乾燥劑乾燥有機相,濃縮後用二氯甲烷重結晶得到無色液體12.2g,產率77%。
如圖6所示,1H NMR(CDCl3,100MHz):δ8.08-8.01(m,2H),7.58(s,1H),7.53-7.39(m,8H),4.13(q,2H,J=7.2Hz),2.74(s,3H),1.01(t,3H,J=7.2Hz)。
實施例7
在500mL的圓底燒瓶中,加入20.0g(50mmol)2-乙醯基-5-氧雜-3,5-二苯基戊酸苄酯,攪拌使其溶解於300mL二甲基亞碸中。將23g(300mmol)醋酸銨固體加入到上述混合液中,室溫攪拌使其溶解。待醋酸銨完全溶解後,將反應體系升至90℃攪拌9小時,並用薄層層析板檢測反應進度。
反應結束,停止攪拌並將反應液降至室溫,加入500mL乙酸乙酯稀釋反應液,並加入蒸餾水洗滌乙酸乙酯層3-5次,每次用水250mL。洗滌結束加入乾燥劑乾燥有機相,濃縮後用二氯甲烷重結晶得到無色液體15.0g,產率79%。
如圖7所示,1H NMR(CDCl3,400MHz):δ8.01(d,2H,J=7.2Hz),7.56(s,1H),7.50-7.36(m,8H),7.25-7.21(m,3H),7.02-6.96(m,2H),5.07(s,2H),2.70(s,2H)。