四氫苯並噻吩衍生物及其製備方法和應用
2024-04-16 06:50:05 3
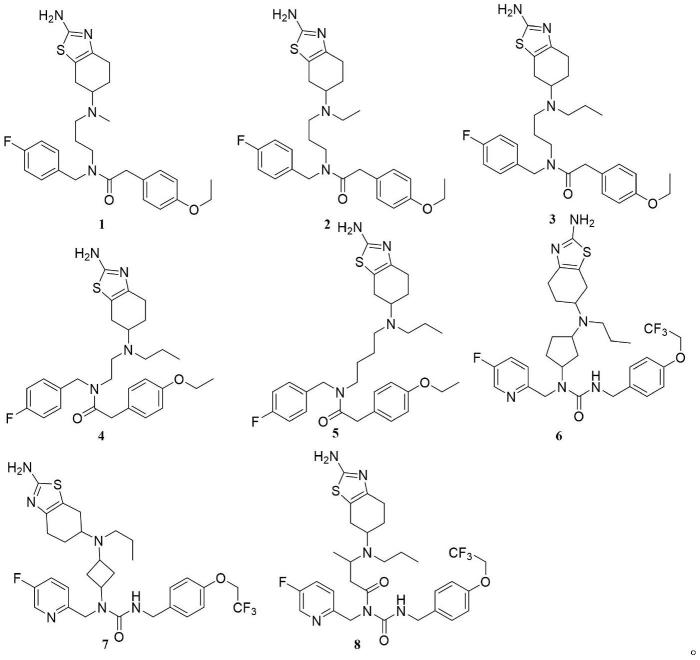
1.本發明涉及醫藥技術領域,具體涉及一類四氫苯並噻吩衍生物及其製備方法和用途,更具體地涉及式(i)化合物及其在製備用於預防或治療帕金森病(pd)患者的帕金森病精神病性障礙(pdp)症狀藥物中的應用。
背景技術:
2.帕金森病是一種常見的中老年神經系統退行性疾病,主要以黑質多巴胺能神經元進行性退變和路易小體形成的病理變化,紋狀體區多巴胺遞質降低、多巴胺與乙醯膽鹼遞質失平衡的生化改變,震顫、肌強直、動作遲緩、姿勢平衡障礙的運動症狀和嗅覺減退、便秘、睡眠行為異常和抑鬱等精神性症狀的臨床表現為顯著特徵。其中約30%~80%帕金森患者會伴發妄想、幻覺等精神症狀。
3.40多年來左旋多巴(l-dopa)一直被認為是治療pd的最有效的藥物,它對各種pd症狀都有較好的療效,但長期和/或大量應用易出現療效減退、「劑末現象」、「開關現象」等併發症的發生。多巴胺受體(d1和d2受體)激動劑可以克服l-dopa的不足,增強l-dopa的療效並延緩併發症的發生。多巴胺受體激動劑早期與複方多巴合用,不僅能提高療效減少複方多巴的用量,而且可減少或避免症狀波動或運動障礙的發生,對疾病後期用複方多巴治療已產生症狀波動或運動障礙者,加用多巴胺受體激動劑的同時減少複方多巴的用量,亦可減輕或消除這些症狀。在疾病後期因黑質紋狀體多巴胺能受體不能把外源性l-dopa脫羧轉化為多巴胺,此時用複方多巴胺完全無效,而用多巴胺受體激動劑則有效。多巴胺受體激動劑已成為目前發展最快的pd治療藥物,如普拉克索、羅替戈汀、羅匹尼羅等。
4.既往認為,多巴胺替代療法與抗精神病治療是一對矛盾,一種症狀的改善可能導致另一種症狀的惡化,治療中遵循的原則是儘可能用最少的多巴胺藥物控制運動症狀,用最低的抗精神病藥物劑量控制精神病性症狀。5-ht
2a
r反向受體激動劑匹莫範色林(pimavanserin)是唯一一個fda批准治療帕金森病精神病性障礙的pdp藥物,可在改善幻覺、妄想等精神症狀的同時不導致運動症狀的惡化。同時,pimavanserin正在探索更為廣泛的應用範圍(痴呆症的精神行為症狀和精神分裂症陰性症狀)。
技術實現要素:
5.本發明提供了式(i)所示的四氫苯並噻唑衍生物、其藥學上可接受的立體異構體、鹽或溶劑合物:
[0006][0007]
其中:r1:選自h、d、c
1-c3取代/未取代烷基、c
1-c3取代/未取代烯基、c
1-c3取代/未取代炔基、烷醯基、苯基烷基、苯基烷醯基;
[0008]
n:選自1、2和3;
[0009]
x1、x2、x3和x4:各自獨立選自cr3、n;
[0010]
r2:選自氫原子、c
1-c
10
直鏈或支鏈的烷基、滷素;
[0011]
l1:選自c
1-c6烷基、c
3-c6環烷基、c
1-c4羰烷基,上述烷基、環烷基、烷羰基可被任意個ra取代;
[0012]
ra:選自h、d、滷素、c
1-c4烷基、c
1-c4滷代烷基、c
1-c6環烷基、c
1-c3烯烴、c
1-c3烷羰基;
[0013]
x:選自c
1-c4烷基、胺基、胺烷基(c
1-c3);
[0014]
r3:選自取代或未取代的苯環/吡啶環/嘧啶環/噠嗪環/三嗪;所述取代基包括:h、d、氨基、羥基、-od、滷素、氰基、硝基、未取代或取代的c
1-6
烷基、未取代或取代的c
2-6
滷代烷基、未取代或取代的c
2-6
炔基、未取代或取代的c
2-6
炔氧基、未取代或取代的烷氧基、未取代或取代的c
3-6
環烷基、未取代或取代的c
3-6
雜脂環基、取代或未取代的芳基、以及取代或未取代的雜芳基,或者取代基及其相連的碳原子、與相連的碳原子一起形成環系,所述環系選自二氫呋喃、二氫吡咯、二氫噻吩,並且被一個或多個相同的或不同的rb取代,每個rb各自獨立地選自h、滷素、c
1-c
10
直鏈或支鏈地烷氧基、c
1-c
10
直鏈或支鏈地滷代烷氧基,其中所述的c1-c10直鏈或支鏈的烷基、c
1-c
10
直鏈或支鏈的烷氧基被選自h、oh、c
1-c
10
直鏈或支鏈的烷氧基中的一個或多個取代基所取代。
[0015]
優選,本發明提供了如下所示化合物或其藥學上可接受的鹽、氘代類似物:
[0016][0017]
再者,涉及式(i)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或溶劑合物的製備方法,其中包括以下步驟:
[0018]
路線1:
[0019][0020]
路線2:
[0021][0022]
路線3:
[0023][0024]
再者,本發明提供了一種藥物組合物,包括治療有效量的上述任一所述化合物或其立體異構體或其藥學上可接受的鹽,或上述任一所述化合物的結晶形式以及藥學上可接受的載體。所述載體,包括本領域中常規的輔料成分,例如填充劑、粘合劑、稀釋劑、崩解劑、潤滑劑、著色劑、調味劑、抗氧化劑和潤溼劑等。
[0025]
所述藥物組合物可以製備成藥學上可接受的各種劑型,如片劑、膠囊劑、口服液劑、混懸液、顆粒劑、粉劑、微粒劑、丸劑、微型片劑、速溶膜劑、鼻噴霧劑、透皮貼劑、注射劑或各種緩控釋製劑等。所述藥物組合物可以經口服、經黏膜、經直腸或腸胃外(包括血管內、靜脈內、腹膜內、皮下、肌肉內和胸骨內)給藥。給藥劑量可根據患者的年齡、性別和疾病類型進行適當調整。
[0026]
對於口服給藥,所述藥物組合物可呈例如片劑、膠囊、液體膠囊、懸浮液或液體形式,所述藥物組合物優選以含有特定量活性成分的劑量單位形式製得。例如,所述藥物組合物可以含有約0.1至1000mg,優選約0.25至250mg,且更優選約0.5至100mg範圍內的量的活性成分的片劑或膠囊提供。用於人類或其它哺乳動物的適合日劑量可根據患者的病況及其它因素而廣泛變化,但可使用常規方法確定。
[0027]
此外,藥理試驗表明,本發明提供的式(i)化合物同時具有較好的5-ht
2a
受體拮抗活性和較高的d
2l
受體激動活性。
[0028]
本發明涉及式(i)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或溶劑合物,或包括式(i)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或
溶劑合物的藥物組合物在製備治療/預防d2和5-ht
2a
受體相關疾病的藥物中的應用。
[0029]
所述疾病或症狀包括帕金森病、精神分裂症、精神病、分裂情感性障礙、雙相性精神障礙、高血壓繼發的精神病、偏頭痛、高血壓、血栓形成、血管痙攣、局部缺血、運動性抽搐、抑鬱、重度抑鬱症、焦慮、睡眠紊亂和食慾紊亂、帕金森病引起的非運動症狀、妄想、幻覺、抑鬱、焦慮、認知障礙、睡眠障礙、痴呆相關的精神疾病、精神分裂的陰性症狀、亨廷頓舞蹈病、阿爾茲海默症、脊髓小腦萎縮症、圖雷特氏症候群、馬查多-約瑟夫病、路易替痴呆、運動障礙、肌陣攣、震顫、進行性核上病麻痺,或其它對本領域技術人員而言顯而易見的其他疾病狀態和狀況。
[0030]
優選,本發明提供的式(ⅰ)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或溶劑合物,或包括式(ⅰ)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或溶劑合物的藥物組合物製備治療帕金森病的運動症狀、並預防/改善精神症狀的藥物的應用,具體的為在製備具有多巴胺受體激動活性、5-羥色胺5-ht
2a
r拮抗活性藥物的應用,也可以為在製備多巴胺受體激動劑和/或5-羥色胺5-ht
2a
r拮抗劑藥物的應用。
[0031]
本發明涉及式(i)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或溶劑合物在製備通過保持d
2l
受體激動作用以治療帕金森的運動症狀藥物的應用,以及在製備通過 5-ht
2a
拮抗作用預防/改善帕金森精神病性症狀藥物的應用。
[0032]
本發明提供了一種新型四氫苯並噻吩衍生物及其製備方法和應用。而且,藥理試驗表明,本發明提供的式(i)化合物同時具有較好的5-ht
2a
受體拮抗活性和較高的d
2l
受體激動活性。本發明提供的式(ⅰ)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或溶劑合物,或包括式(ⅰ)所示的四氫苯並噻吩衍生物或其立體異構體、藥學可接受的鹽或溶劑合物的藥物組合物在d2受體激動作用的基礎上,通過增加5-ht
2a
r拮抗作用預防/改善精神病性症狀,為新一代pd藥物研發奠定基礎,並有望在未來造福廣大pd患者。
[0033]
定義和說明
[0034]
除非另有說明,本文所用的下列術語和短語旨在具有下列含義。一個特定的術語或短語在沒有特別定義的情況下不應該被認為是不確定的或不清楚的,而應該按照普通的含義去理解。當本文中出現商品名時,意在指代其對應的商品或其活性成分。
[0035]
這裡所採用的術語「藥學上可接受的」,是針對那些化合物、材料、組合物和/或劑型而言,它們在可靠的醫學判斷的範圍之內,適用於與人類和動物的組織接觸使用,而沒有過多的毒性、刺激性、過敏性反應或其它問題或併發症,與合理的利益/風險比相稱。
[0036]
術語「藥學上可接受的鹽」是指本發明化合物的鹽,由本發明發現的具有特定取代基的化合物與相對無毒的酸或鹼製備。當本發明的化合物中含有相對酸性的功能團時,可以通過在純的溶液或合適的惰性溶劑中用足夠量的鹼與這類化合物的中性形式接觸的方式獲得鹼加成鹽。當本發明的化合物中含有相對鹼性的官能團時,可以通過在純的溶液或合適的惰性溶劑中用足夠量的酸與這類化合物的中性形式接觸的方式獲得酸加成鹽。藥學上可接受的酸加成鹽的實例包括無機酸鹽、有機酸鹽、還包括胺基酸(如精氨酸等)的鹽,以及葡糖醛酸等有機酸的鹽。本發明的某些特定的化合物含有鹼性和酸性的官能團,從而可以被轉換成任一鹼或酸加成鹽。
[0037]
本發明的藥學上可接受的鹽可由含有酸根或鹼基的母體化合物通過常規化學方法合成,一般情況下,鹽的製備方法為:在水或有機溶劑或兩者的混合物中,經由游離酸或
鹼形式的這些化合物與化學計量的適當的鹼或酸反應來製備。
[0038]
本發明的某些化合物可以具有不對稱碳原子(光學中心)或雙鍵、外消旋體、非對映異構體、幾何異構體和單個的異構體都包括在本發明的範圍之內。
[0039]
本發明的化合物可以存在特定的幾何或立體異構體形式。本發明設想所有的這類化合物,包括順式和反式異構體,(-)-和(+)-對對映體、r和s對映體、非對映異構體、d型、l型異構體,及其外消旋混合物和其他混合物,例如對映異構體或非對映體富集的混合物。所有這些異構體以及它們的混合物,均包括在本發明的範圍之內。
[0040]
可以通過手性合成或手性試劑或其他常規技術製備光學活性的r型和s型異構體以及d型和l型異構體。如果想得到本發明某化合物的一種對映體,可以通過不對稱合成或者具有手性助劑的衍生作用來製備,其中將所得非對映體混合物分離,並且輔助基團裂開以提供純的所需對映異構體。或者,當分子中含有鹼性官能團(如氨基)或酸性官能團(如羧基)時,與適當的光學活性的酸或鹼形成非對映異構體的鹽,然後通過本領域所公知的常規方法進行非對映異構體拆分,然後回收得到純的異構體。此外,對映異構體和非對映異構體的分離通常是通過使用色譜法完成的,所述色譜法採用手性固定相,並任選地與化學衍生法相結合(例如由胺生成氨基甲酸鹽)。
[0041]
術語「藥學上可接受的載體」是指能夠遞送本發明有效量活性物質、不幹擾活性物質的生物活性並且對宿主或者患者無毒副作用的任何製劑或載體介質代表性的載體,包括但不限於:粘合劑、填充劑、潤滑劑、崩解劑、潤溼劑、分散劑、增溶劑、助懸劑等。
[0042]
針對藥物或藥理學活性劑而言,術語「有效量」或「治療有效量」是指無毒的但能達到預期效果的藥物或藥劑的足夠用量。有效量的確定因人而異,取決於受體的年齡和一般情況,也取決於具體的活性物質,個案中合適的有效量可以由本領域技術人員根據常規試驗確定。
[0043]
本發明意欲包括存在於本發明化合物中的原子的所有同位素。同位素包括原子數相同但質量數不同的那些原子。作為一般實例且非限制性地,氫的同位素包括氘和氚。碳的同位素包括
13
c和
14
c。本發明的同位素標記化合物一般可通過本領域技術人員已知的常規技術或通過類似於本文所述的方法,使用適當同位素標記試劑代替另外使用的非標記試劑來製備。
[0044]
術語「氘代類似物」指的是化合物的一個或多個氫原子被氘原子替代所產生的類似物。術語「任選」或「任選地」指的是隨後描述的事件或狀況可能但不是必需出現的,並且該描述包括其中所述事件或狀況發生的情況以及所述事件或狀況下不發生的情況。例如,「任選地被一個或多個氘原子取代」是指所述基團可以是未被氘原子取代的情況,或者被一個或多個氘原子取代的情況,即保護了基團未被氘代、部分氘代和/或全部氘代的情況。
[0045]
術語「被取代的」是指特定原子上的任意一個或多個氫原子被取代基取代,可以包括重氫和氫的變體,只要特定原子的價態是正常的並且取代後的化合物是穩定的。當取代基為酮基時,意味著兩個氫原子被取代。酮取代不會發生在芳香基上。
[0046]
當任何變量(例如r)在化合物的組成或結構中出現一次以上時,其在每一種情況下的定義都是獨立的。因此,例如,如果一個基團被0-2個r所取代,則所述基團可以任選地至多被兩個r所取代,並且每種情況下的r都有獨立的選項。此外,取代基和/或其變體的組合只有在這樣的組合會產生穩定的化合物的情況下才是被允許的。
[0047]
除非另有規定,術語「烷基」用於表示直鏈或支鏈的飽和烴基,可以是單取代(如-ch2f) 或多取代的(-cf3),可以是一價(如甲基)、二價(如亞甲基)或者多價(如次甲基)。例如如c
1-c
10
表示1-10個碳,c
1-c
10
選自c1、c2、c3、c4、c5、c6、c7、c8、c9和c
10
;烷基的例子包括甲基(me),乙基(et),丙基(如n-丙基和異丙基),丁基(如n-丁基,異丁基,s-丁基, t-丁基),戊基(如n-戊基,異戊基,新戊基,1-乙基丙基),己基(如n-己基,異己基,1,1
‑ꢀ
二甲基乙基,2,2-二甲基丁基,3,3-二甲基丁基,2-乙基丁基),庚基,辛基,壬基,癸基等。
[0048]
除非另有規定,術語「滷代素」或「滷素」本身或作為另一取代基的一部分表示氟、氯、溴或碘原子。術語「滷代烷氧基」意在包括單滷代烷基和多滷代直鏈或支鏈的滷代烷氧基。例如,術語「滷代c
1-c
10
烷氧基」意在包括但不僅限於氟甲氧基、二氟甲氧基、三氟甲氧基、三氯甲氧基、2-氟乙氧基、2,2-氟乙氧基、2,2,2-三氟乙氧基、四氟乙氧基、五氟乙氧基、3
‑ꢀ
氟丙氧基、3,3-二氟丙氧基、2,2
』‑
二氟異丙氧基、3,3,3-三氟丙氧基、4-氟丁氧基、4,4-二氟丁氧基、4,4,4-三氟丁氧基、2-氟-2甲基丙基、5,5,5-三氟戊氧基、6,6,6-三氟己氧基。
[0049]「滷代烷基」意在包括單滷代烷基和多滷代直鏈或支鏈的烷基。例如,術語「滷代(c
1-c4) 烷基」意在包括但不僅限於三氟甲基、2,2,2-三氟乙基、4-氯丁基和3-溴丙基等等。除非另有規定,滷代烷基的實例包括但不僅限於:三氟甲基、三氯甲基、五氟乙基和五氯乙基。
[0050]
除非另有規定,「烷氧基」代表通過氧橋連接的具有特定數目碳原子的上述烷基。典型的烷氧基包括c
1-c
10
烷氧基,例如c1、c2、c3、c4、c5、c6、c7、c8、c9、c
10
的烷氧基。烷氧基的例子包括但不限於:甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、仲丁氧基、叔丁氧基、正戊氧基、s-戊氧基、己氧基、2-乙基丁氧基、庚氧基、辛氧基、壬氧基、癸氧基等。
[0051]
除非另有規定,環烷基包括任何穩定的環狀或多環烴基,任何碳原子都是飽和的,可以是單取代或多取代的,可以是一價、二價或者多價。這些環烷基的實例包括,但不限於,環丙基、環丁基、環戊基、環己基、環庚基、環辛基、降冰片烷基、[2,2,2]二環辛烷、[4,4,0] 二環癸烷等。
具體實施方式
[0052]
下面將結合具體實施例對本發明進行清楚、完整的描述,本領域技術人員將會理解,下述實施例是本發明部分實施方式,而不是全部實施方式,僅用於說明本發明,而不應視為對本發明保護範圍的限制。
[0053]
本發明中,未註明具體試驗條件的,按照常規試驗條件或製造商建議的條件進行,所用試劑或儀器未註明生產廠商者,均可以通過市售購買獲得常規產品。
[0054]
本發明中,試驗結果以平均值表示。
[0055]
本發明中的檢測指標為:人d
2l
受體親和力試驗;人5-ht
2a
受體親和力試驗;人d
2l
受體功能試驗(激動劑檢測);人5-ht
2a
受體功能試驗(拮抗劑檢測)。
[0056]
實施例1
[0057][0058]
合成路線:
[0059][0060]
1)化合物1-2的合成
[0061]
將乙醇胺(20g,266.28mmol,20.53ml,1eq)、化合物1-1(34.70g,279.59mmol,29.41ml, 1.05eq)、醋酸(1.60g,26.63mmol,1.52ml,0.1eq)和甲醇(200ml)加入至500ml配有溫度計的三口瓶,20℃攪拌2小時。醋酸硼氫化鈉(62.08g,292.90mmol,1.1eq)加入至反應瓶,20℃攪拌16小時,反應過程中體系混濁。過濾,濾液濃縮幹得到粗品。粗品經柱層析純化得到純品化合物1-2(黃色油狀物,34.1g,收率:69.89%,純度:100%)。ms(esi)m/z:184.3[m+h]
+
。1h nmr(400mhz,cdcl3)δ7.27
–
7.25(m,2h),7.02
–
6.98(m,2h),3.80(t,j=10.8hz,2h), 3.75(s,2h),2.88(t,j=11.6hz,2h),2.41(br,1h),1.75
–
1.69(m,2h)。
[0062]
2)化合物1-3的合成
[0063]
將二氯甲烷(350ml)、4-乙基氧丙基乙酸酯(7.01g,1.02eq)、edci(8.78g,1.2eq),hobt (6.20g,1.2eq)和dipea(7.80g,1.58eq)依次加入至500ml配有溫度計的三口瓶中,冰水浴 0~5℃反應1小時後,將化合物1-2(7.00g,1eq)分批加入至反應液中,20℃反應16小時。反應液用飽和碳酸氫鈉溶液調節ph至7~8,靜置,分液。有機相用無水硫酸鈉乾燥,過濾,濾液濃縮幹,得到粗品,粗品經柱層析純化後得到純品化合物1-3(黃色油狀物,6.00g,收率: 45.5%)。ms(esi)m/z:346.2[m+h]
+
。1h nmr(400mhz,cdcl3)δ7.18-7.13(m,2h),7.07
‑ꢀ
7.02(m,4h),6.85-6.83(m,2h),4.46(s,2h),4.02(q,j=6.8hz,2h),3.84-3.82(m,1h),3.66(s, 2h),3.53-3.46(m,4h),1.6-1.58(m,2h),1.40(t,j=8.0hz,3h)。
[0064]
3)化合物1-4的合成
[0065]
將10ml無水二氯甲烷、草醯氯(440.96mg,3.47mmol,304.11ul,1.2eq)依次加入至100 ml配有溫度計的三口瓶,用乾冰乙醇體系降溫至-65~-70℃,氮氣保護下攪拌0.5小
eq)加入至反應體系,20℃保溫攪拌16小時。向反應體系中加入60ml水,混合體系用二氯甲烷萃取2次,每次50ml。有機相合併用無水硫酸鈉乾燥,過濾,濾液濃縮幹得到粗品。粗品經製備色譜分離純化得到實施例2(白色固體,390mg,收率:26.1%,純度:99%)。ms(esi) m/z:525.3[m+h]
+
。1h nmr(400mhz,dmso-d6)δ7.30-7.06(m,6h),6.85-6.75(m,2h), 6.60(s,2h),4.55(s,1h),4.45(s,1h),4.05-3.94(m,2h),3.65(s,1h),3.60(s,1h),3.30-3.20(m, 2h),2.95-2.80(m,1h),2.45-2.30(m,8h),1.80-1.65(m,1h),1.50-1.45(m,3h),0.94(t,j= 6.0hz,3h)。
[0073]
7)化合物實施例3的合成
[0074]
參考實施例1中的合成方法,將40ml甲醇、化合物1-5(2.6g,5.24mmol,1eq)、丙醛(1.24 g,28.19mmol,1.58ml,10eq)、醋酸(31.44mg,523.52umol,29.94ul,0.1eq)依次加入至 100ml配有溫度計的三口瓶,20℃攪拌半小時後,將醋酸硼氫化鈉(1.66g,7.85mmol,1.5eq) 加入至反應體系,20℃保溫攪拌16小時。向反應體系中加入50ml水,混合體系用二氯甲烷萃取2次,每次50ml.有機相合併用無水硫酸鈉乾燥,過濾,濾液濃縮幹得到粗品。粗品經製備色譜分離純化得到實施例3(白色固體,370mg,收率:12.5%,純度:95.1%)。ms(esi)m/z: 539.4[m+h]
+
。1h nmr(400mhz,dmso-d6)δ7.30-7.05(m,6h),6.85-6.75(m,2h),6.55(s, 2h),4.55(s,1h),4.45(s,1h),4.00-3.90(m,2h),3.65(s,1h),3.60(s,1h),3.30-3.15(m,2h), 2.90-2.75(m,1h),2.45-2.30(m,8h),1.75-1.60(m,1h),1.50-1.45(m,3h),1.35-1.20(m, 5h),0.78(t,j=6.0hz,3h)。
[0075]
實施例4
[0076][0077]
合成路線:
[0078][0079]
1)化合物4-3的合成
[0080]
將400ml甲醇、化合物4-1(19.6g,19.4ml,1.05eq)、化合物4-2(38.0g,32.2ml,1.00eq) 依次加入至1l配有溫度計的三口瓶,室溫攪拌2小時後,將醋酸硼氫化鈉(71.4g,1.10eq) 加入至反應液,室溫下攪拌12小時。向反應液中加入150ml水,過濾,濃縮除去甲醇,水相用乙酸乙酯萃取三次,每次200ml。有機相干燥,過濾,濾液濃縮幹得到化合物4-3(黃色油狀物,51.8g)。1h nmr:(400mhz,cdcl3)δ=9.95-9.88(m,1h),8.22(s,1h),7.71-7.62(m, 2h),7.12-6.99(m,2h),3.91-3.79(m,2h),3.74-3.65(m,2h),3.43-3.32(m,3h),3.22-3.14 (m,1h),2.99(brs,3h),1.16(s,3h)。
[0081]
2)化合物4-4的合成
[0082]
將250ml無水thf、化合物4-3(26.6g,1.00eq)、boc2o(27.5g,0.80eq)、催化量的dmap 依次加入到500ml三口瓶,室溫下攪拌3小時。向反應液中加入250ml水,200ml乙酸乙酯萃取一次,有機相干燥,過濾,濃縮得到粗品。粗品經柱層析純化得到化合物4-4(無色油狀,26.0g,收率59.2%,純度96.4%)。ms(esi)m/z:[m+h]
+
。1h nmr(400mhz,cdcl3)δ7.21 (s,2h),7.02(t,j=8.6hz,2h),4.45(s,2h),3.70(s,2h),3.39(s,2h),2.22(s,1h),1.47(s,9h)。
[0083]
3)化合物4-5的合成
[0084]
將無水100ml二氯甲烷、草醯氯(5.66g,1.20eq)依次加入到500ml配有低溫溫度計的三口瓶,降溫至-70℃,攪拌30分鐘。將二甲基亞碸(4.35g,4.35ml,1.50eq)溶於10ml無水二氯甲烷,逐滴加入反應液,溫度控制在-60~-70℃。加畢後,-70℃保溫攪拌1小時。將化合物4-4(10.0g,1.00eq)溶於20ml無水二氯甲烷,控制溫度在-60~-70℃,滴加到反應液中,保溫攪拌2小時。在-60~-70℃下,逐滴滴加diea(24.0g,32.3ml,5.00eq),加畢後,保溫攪拌30分鐘。升至室溫後,保溫攪拌16小時。將反應液倒入150ml水中,分離有機相,有機相用1m檸檬酸水溶液洗滌兩次,每次150ml;150ml碳酸氫鈉水溶液洗滌一次,無水硫酸鈉乾燥,過濾,濾液濃縮幹得到粗品。粗品經柱層析純化得到化合物4-5(黃色油狀,6.30g)。 ms(esi)m/z:212.1[m+h]
+
。1h nmr(400mhz,cdcl3)δ9.51-9.38(m,1h),7.25-7.12(m, 2h),7.01(t,j=8.4hz,2h),4.47(d,j=19.1hz,2h),3.99-3.71(m,2h),2.03(s,1h),1.70
‑ꢀ
1.33(m,11h)。
[0085]
4)化合物4-6的合成
[0086]
將100ml甲醇、化合物4-5(5.30g,1.00eq)、4,5,6,7-四氫-2,6-苯並噻唑胺(332.69mg,1.97 mmol,1eq)、醋酸(595mg,0.50eq)依次加入至250ml三口瓶,室溫攪拌30分鐘,將氰基硼氫化鈉(1.87g,1.50eq)加入至反應液中,室溫攪拌2小時。向反應液中加入150ml,二氯甲烷萃取兩次,每次150ml。合併有機相,用無水硫酸鈉乾燥,過濾,旋幹得到粗品,粗品經柱層析純化得到化合物4-6(黃色固體,3.80g)。ms(esi)m/z:421.1[m+h]
+
。
[0087]
5)化合物4-7的合成
[0088]
將40.0ml甲醇、化合物4-6(1.00g,1.00eq)、丙醛(414mg,519ul,3.00eq)、醋酸(14.3 mg,13.6ul,0.10eq)依次加入到100ml單口瓶,室溫下攪拌30分鐘。將氰基硼氫化鈉加入到反應液中,室溫攪拌30分鐘。向反應液中加入10ml水,濃縮除去甲醇後,用二氯甲烷萃取兩次,每次45ml,合併有機相,用無水硫酸鈉乾燥,過濾,濃縮得到粗品,粗品經液相製備純化得到化合物4-7(黃色油狀物,1.38g)。ms(esi)m/z:463.1[m+h]
+
。
[0089]
6)化合物4-8的合成
[0090]
將15ml二氯甲烷、化合物4-7(1.00g,1.00eq)、三氟乙酸(3.70g,15.0eq)依次加入到 50ml單口反應瓶中,室溫攪拌20小時。向反應液中緩慢加入30ml飽和碳酸氫鈉水溶液,用二氯甲烷萃取三次,每次30ml,合併有機相,用無水硫酸鈉乾燥,過濾,濃縮得到化合物4-8 (黃色固體,540mg,收率66.2%,純度96.0%)。ms(esi):363.2[m+h]
+
。1h nmr(400mhz, dmso-d6)δ7.44-7.36(m,2h),7.23-7.11(m,2h),6.64-6.56(m,2h),3.88-3.79(m,2h),2.93
ꢀ‑
2.84(m,1h),2.54(d,j=5.8hz,4h),2.49-2.45(m,1h),2.45-2.31(m,4h),1.86-1.78(m, 1h),1.65-1.52(m,1h),1.41-1.30(m,2h),0.81(t,j=7.3hz,3h)。
[0091]
7)化合物實施例4的合成
[0092]
將40ml無水二氯甲烷、4-乙基氧丙基乙酸酯(656.2mg,1.10eq)、hobt(536.8mg,1.20 eq)、edci(761.5mg,1.20eq)、三乙胺(1.00g,1.38ml,3.00eq)依次加入到100ml配有溫度計的三口瓶,0℃保溫攪拌30分鐘後,將化合物4-8(1.20g,1.00eq)加入至反應液中,升至室溫攪拌3.5小時。向反應液中加入60ml飽和氯化銨水溶液,用40ml二氯甲烷萃取。有機相干燥,過濾,濃縮得到粗品。粗品經製備色譜純化得到實施例4(黃色膠狀物,1.07g,收率62.0%,純度98.0%)。ms(esi)m/z:525.2[m+h]
+
。1h nmr(400mhz,dmso-d6)δ7.28
‑ꢀ
7.02(m,6h),6.89-6.75(m,2h),6.34-6.20(m,2h),4.66-4.50(m,2h),4.05-3.94(m,2h), 3.80-3.55(m,2h),3.31-3.21(m,2h),2.90-2.81(m,1h),2.58-2.51(m,2h),2.44-2.29(m, 4h),1.82-1.73(m,1h),1.59-1.47(m,1h),1.38-1.25(m,5h),0.85-0.75(m,3h)。
[0093]
實施例5
[0094][0095]
合成路線:
[0096][0097]
1)化合物5-2的合成
[0098]
將130ml無水甲醇、化合物5-1(13g,1eq)化合物4-氨基-1-丁醇(9.80g,1.05eq)依次加入250ml單口瓶,室溫攪拌2小時後分批次加入醋酸硼氫化鈉(24.42g,1.1eq),室溫攪拌16小時。向反應液中加入50ml水,濃縮除去甲醇。用乙酸乙酯萃取三次,每次100ml,合併有機相,用無水硫酸鈉乾燥,過濾,濃縮幹,得到粗品中間體5-2(黃色油狀物,20.66g)。
[0099]
2)化合物5-3的合成
[0100]
將26ml無水dmf、化合物4-乙基氧丙基乙酸酯(1.57g,1eq)、hobt(1.41g,1.2eq)、 edci(2.01g,1.2eq)、diea(1.69g,1.5eq)依次加入100ml單口瓶,0℃小攪拌1小時,將化合物2(2.58g,1.5eq)加入至反應液,0~10℃攪拌2小時。向反應液中加入30ml水,乙酸乙酯萃取兩次,每次50ml。合併有機相,有機相干燥,過濾,濃縮幹得到粗品,粗品經柱層析純化得到化合物5-3(黃色油狀物,1.87g,收率:58.77%,純度:98.5%)。ms(esi)m/z: =360.2[m+h]
+
。
[0101]
3)化合物5-4的合成
[0102]
將16ml無水二氯甲烷、草醯氯(665.29mg,1.2eq)加入至100ml配有溫度計的三口瓶,降溫至-70℃,攪拌30分鐘。將二甲基亞碸(511.93mg,1.5eq)溶於2ml無水二氯甲烷,逐滴加入至反應液中,控制溫度在-70℃左右,加畢後,-70℃攪拌1小時。將化合物5-3(1.57g, 1eq)溶於3ml無水二氯甲烷,緩慢滴加到反應液中,維持溫度在-70℃左右。加畢後保溫攪拌2小時。將diea(2.82g,21.84mmol,3.80ml,5eq)逐滴加入至反應液中,控制溫度在-70℃左右,保溫攪拌30分鐘,升溫至室溫,攪拌2小時。反應液倒入至20ml水,分離有機相,有機相分別用40ml10%檸檬酸溶液、20ml飽和碳酸氫鈉溶液洗滌一次,用無水硫酸鈉乾燥,過濾,濃縮得到化合物5-4(黃色油狀物,1.49g,收率:87.50%,純度:92.2%)。ms(esi)m/z: 358.1[m+h]
+
。
[0103]
4)化合物5-5的合成
[0104]
將6ml甲醇、化合物5-4(600mg,1eq)、4,5,6,7-四氫-2,6-苯並噻唑胺(284.12mg,1eq)依次加入到50ml三口瓶中,室溫下攪拌3小時。將氰基硼氫化鈉(158.24mg,1.5eq)加入到反應液中,室溫攪拌19小時。向反應液中加入6ml水,用乙酸乙酯萃取兩次,每次10ml。合
並有機相,有機相用無水硫酸鈉乾燥,過濾,濃縮幹,得到粗品。粗品經柱層析純化得到化合物5-5(黃色油狀,440mg,收率:51.33%)。ms(esi)m/z:511.2[m+h]
+
。
[0105]
5)化合物實施例5的合成
[0106]
將5ml甲醇、化合物5-5(440mg,1eq)、丙醛(750.63mg,940.64ul,15eq)依次加入到 10ml反應瓶,室溫攪拌2小時。將氰基硼氫化鈉(81.22mg,1.5eq)加入反應液,室溫下攪拌16小時。向反應液中加入5ml水,用乙酸乙酯萃取兩次,每次10ml。合併有機相,用無水硫酸鈉乾燥,過濾,濃縮,得到粗品。粗品經製備色譜分離得到實施例5(無色膠狀,130 mg,收率:27.08%,純度:99.2%)。ms(esi)m/z:511.2[m+h]
+
。1h nmr(400mhz,cdcl3) δ7.20-6.95(m,6h),6.89-6.75(m,2h),4.73(s,2h),4.56(s,2h),4.47(s,2h),4.05(q,j=6.8 hz,2h),3.71(s,h),3.62(s,1h),3.33(t,j=7.60,1h),3.18(t,j=7.60hz,1h),3.05-2.95(m, 1h),2.75-2.30(m,8h),2.01-1.85(m,1h),1.75-1.25(m,10h),0.90-0.75(m,3h)。
[0107]
實施例6
[0108][0109]
合成路線:
[0110][0111]
1)化合物6-2的合成
[0112]
將60ml甲醇、化合物6-1(4.01g,19.0mmol,1.00eq)、3-(boc-氨基)環戊酮(3.78g,19.0 mmol,1.00eq)、氰基硼氫化鈉(1.79g,28.5mmol,1.50eq)、氯化鋅(259mg,1.90mmol,88.9 ul,0.10eq)依次加入到250ml三口瓶,升溫到50~60℃,攪拌30小時。向反應液中加入30ml 水,用30ml乙酸乙酯萃取,有機相用無水硫酸鈉乾燥,濃縮得到粗品。粗品經柱層析純化得到化合物6-2(棕色油狀物,2.20g,4.83mmol,收率:25.5%,純度:86.7%)。ms(esi)m/
z:395.3 [m+h]
+
。1h nmr(400mhz,methanol-d4)δ4.07-3.94(m,1h),3.89(td,j=7.1,14.5hz, 1h),2.96(tdd,j=2.6,8.6,13.3hz,1h),2.85(br dd,j=4.4,15.3hz,1h),2.58-2.51(m,1h), 2.49-2.41(m,1h),2.41-2.32(m,1h),2.13-2.03(m,2h),1.98-1.83(m,2h),1.66(ddd,j=5.9, 10.8,12.5hz,2h),1.56(br dd,j=7.6,15.2hz,3h),1.46(brs,2h),1.43(s,10h),0.96(t,j=7.4 hz,3h)。
[0113]
2)化合物6-3的合成
[0114]
將23ml二氯甲烷、中間體6-2(2.20g,5.58mmol,1.00eq)、三氟乙酸(6.36g,55.8mmol, 4.13ml,10.0eq)依次加入至100ml單口瓶中,室溫下攪拌12小時。減壓濃縮除去溶劑,向反應液中加入碳酸鉀溶液,調節ph至7
–
8,用二氯甲烷萃取三次,每次50ml,合併有機相,乾燥,濃縮得到粗品。粗品經柱層析純化得到化合物6-3(黃色固體物,3.70g)。ms(esi) m/z:295.1[m+h]
+
。
[0115]
3)化合物6-4的合成
[0116]
將34ml甲醇、化合物6-3(3.70g,4.75mmol,37.8%purity,1.00eq)、5-氟吡啶-2-甲醛(535 mg,4.27mmol,0.90eq)依次加入到100ml單口瓶,0℃攪拌2小時。將醋酸硼氫化鈉(1.51 g,7.12mmol,1.50eq)加入到反應液中,0℃攪拌1小時。向反應液中加入10ml水,濃縮除去甲醇,用乙酸乙酯萃取兩次,每次20ml,合併有機相,乾燥,濃縮幹,得到粗品。粗品經柱層析純化得到化合物6-4(黃色油狀物,2.57g)。ms(esi)m/z:390.1[m+h]
+
。
[0117]
4)化合物實施例6的合成
[0118]
將30ml無水thf、(4-(2,2,2-三氟乙氧基)苯基)甲胺(942mg,4.59mmol,0.75eq)加入到 100ml單口瓶,降溫至0℃,加入cdi(1.49g,9.18mmol,1.50eq),保溫攪拌1h後將化合物6-4 (2.47g,6.12mmol,1.00eq)溶於30ml無水thf,40℃保溫攪拌14小時。反應液濃縮得到粗品,粗品經製備色譜分離得到實施例6(黃色固體,440mg,692umol,收率:11.3%,純度:99.8%)。 ms(esi)m/z:635.3[m+h]
+
。1h nmr(400mhz,dmso-d6)δ8.50-8.43(m,1h),7.71-7.61 (m,1h),7.41-7.21(m,1h),7.20-7.14(m,2h),7.08-6.91(m,3h),6.65-6.45(m,2h),4.71(q, j=9.0hz,2h),4.57-4.29(m,3h),4.23-4.15(m,2h),3.15-2.65(m,2h),2.46-2.30(m,5h), 2.03-1.19(m,11h),0.92-0.70(m,3h)。
[0119]
實施例7
[0120][0121]
合成路線:
[0122][0123]
1)化合物7-2的合成
[0124]
將75ml甲醇、化合物1(5.00g,1.00eq)、3-(boc-氨基)-1-環丁酮(4.38g,23.7mmol,1.00 eq)、氯化鋅(3.22g,1.00eq)、醋酸硼氫化鈉(2.23g,1.50eq)依次加入到250ml三口瓶,50℃保溫攪拌12小時。向反應液中加入30.0ml水,用乙酸乙酯萃取兩次,每次15ml,合併有機相,用無水硫酸鈉乾燥,濃縮幹,得到粗品。粗品經柱層析純化得到化合物7-2(棕色固體, 2.60g,收率:28.0%,純度:96.8%)。ms(esi)m/z:381.3[m+h]
+
。1hnmr(400mhz,meoh-d4) δ3.76-3.60(m,1h),3.14-2.97(m,2h),2.64-2.53(m,3h),2.47(dt,j=6.7,13.9hz,5h),1.99
ꢀ‑
1.91(m,1h),1.82(q,j=8.8hz,2h),1.71(br dd,j=5.4,12.1hz,1h),1.56-1.39(m,12h), 0.89(t,j=7.3hz,3h)。
[0125]
2)化合物7-3的合成
[0126]
將28ml二氯甲烷、化合物7-2(2.60g,1.00eq)加入到100ml單口瓶,將三氟乙酸(7.79g, 5.06ml,10.0eq)在0℃滴加至反應液中,室溫下反應12小時。將反應液濃縮幹,用碳酸鉀水溶液調節ph至中性。將混合液濃縮幹得到粗品。粗品經柱層析得到化合物7-3(黃色固體,1.76g)。 ms(esi)m/z:281.2[m+h]
+
。
[0127]
3)化合物7-4的合成
[0128]
將34ml甲醇、化合物7-3(1.36g,1.00eq)、5-氟吡啶-2-甲醛(546mg,0.90eq)加入至100 ml單口瓶,0℃保溫攪拌2小時後,將醋酸硼氫化鈉(1.54g,1.50eq)加入至反應液,0℃保溫攪拌10分鐘。向反應液中加入1ml水,濃縮幹,得到粗品。粗品經柱層析純化得到中間體7-4 (黃色油狀物,1.82g)。ms(esi)m/z:390.1[m+h]
+
。
[0129]
4)化合物實施例7的合成
[0130]
將30ml無水四氫呋喃、(4-(2,2,2-三氟乙氧基)苯基)甲胺(958mg,1.00eq)加入到100ml單口瓶,降溫至0℃後,將cdi(1.14g,1.50eq)加入至反應液,保溫攪拌1小時。將化合物7-4(1.82 g,1.00eq)溶於30.0ml無水四氫呋喃後滴加至反應液中,40℃保溫攪拌14小時。反應液濃縮幹,得到粗品。粗品經製備色譜純化得到實施例7(黃色固體,750mg,收率:25.7%,純度: 99.2%)。ms(esi)m/z:300.1[m+h]
+
。1h nmr(400mhz,dmso-d6)δ8.55-8.44(m,1h),7.77
ꢀ‑
7.59(m,1h),7.32-7.08(m,3h),7.03-6.82(m,3h),6.75-6.50(m,2h),4m,2h),4.10-3.96 (m,1h),3.01-2.76(m,2h),2.46-2.17(m,7h),1.91-1.50(m,4h),1.42-1.04(m,3h),0.77(brs,3h)。
[0131]
實施例8
[0132][0133]
合成路線:
[0134][0135]
1)化合物8-2的合成
[0136]
將240ml四氫呋喃、化合物8-1(30.0g,1.00eq)加入到500ml三口瓶,降溫至0℃,將 boc2o(42.5g,1.10eq)溶於40ml四氫呋喃,0℃下保溫滴加至反應溶液,室溫攪拌12小時。向反應液中加入100ml水,用乙酸乙酯萃取三次,每次100ml。合併有機相,乾燥,過濾,濾液濃縮幹,得到粗品,粗品經柱層析純化得到化合物8-2(白色固體,31.0g,收率:58.4%,純度:90%)。1h nmr(400mhz,cdcl3)δ4.81(brs,1h),4.62(br d,j=5.6hz,1h),3.98(brs,1h), 2.88(br dd,j=4.6,15.8hz,1h),2.66-2.51(m,2h),2.39(br dd,j=6.7,15.8hz,1h),1.91(dtd, j=2.9,6.3,12.8hz,1h),1.77(qd,j=6.9,13.4hz,1h),1.38(s,9h)。
[0137]
2)化合物8-3的合成
[0138]
將310ml二氯甲烷、化合物8-2(31.0g,1.00eq)、diea(29.5g,1.98eq)、cbzcl(20.2g,1.03 eq)依次加入到500ml三口瓶,室溫下保溫攪拌12小時。向反應液中加入200ml水,分液,有機相干燥,過濾,濃縮得到粗品。粗品經柱層析純化得到化合物8-3(白色固體,21g,收率: 45.2%)。1h nmr(400mhz,cdcl3)δ7.43-7.35(m,5h),5.24(s,2h),4.65-4.45(m,1h),3.97 (brs,1h),3.00(br dd,j=4.9,15.8hz,1h),2.61-2.39(m,3h),1.83-1.68(m,1h),1.67-1.51 (m,2h),1.47(s,9h)。
[0139]
3)化合物8-4的合成
[0140]
將300ml二氯甲烷、化合物8-3(18g,1.00eq)加入到500ml單口瓶,將tfa(50.8g,10.0 eq)緩慢滴加到反應液中,室溫下保溫攪拌12小時。向反應液中加入150ml水,分離水相,水相用乙酸乙酯萃取兩次,每次150ml,丟棄有機相,水相用碳酸鈉固體調節ph=9,濃縮
406.2[m+h]
+
。1h nmr(400mhz,cdcl3)δ8.80-8.71(m,1h),8.35(dd,j=2.8,9.8hz,1h), 7.38-7.31(m,1h),4.90(brs,2h),4.60-4.45(m,2h),3.42-3.33(m,1h),3.17-3.03(m,1h), 2.60-2.39(m,6h),2.19(ddd,j=3.6,9.5,15.9hz,1h),2.06-1.95(m,1h),1.90-1.66(m,2h), 1.56-1.24(m,3h),1.06(t,j=6.0hz,3h),0.83(dt,j=3.2,7.3hz,3h)。
[0151]
試驗例1人5-ht2a受體功能試驗(拮抗劑檢測)
[0152]
細胞:穩定表達人源5-ht
2a
的hek293細胞。
[0153]
試劑:dmem(invitrogen);pbs(corning);g418(invitrogen);blasticidin(invitrogen); penicillin/streptomycin(hyclone);fluo-4(invitrogen)。
[0154]
設備:384 well plate,greiner;vi-cell xr cell viability analyzer,beckman coulter;孵育箱, thermo。
[0155]
方法:將hek293細胞以20000/孔密度接種於384孔板,隔天測試化合物使用dmso配置,每孔加入0.9μl,測試濃度為10000nm、2500nm、625nm、156nm、39nm、9.76nm、 2.44nm、0.61nm、0.15nm、0.038nm,然後加入20μl fluo-4 directtm no-wash loading buffer 後放回孵箱,37℃,孵育50min,然後室溫孵育10min。使用flipr tetra高通量實時螢光檢測分析系統測定螢光值。化合物ic
50
、ic
90
使用prism計算得到,結果見下表。
[0156]
化合物編號ic
50
(nm)ic
90
(nm)匹莫範色林12.3620.4實施例116.5131.05實施例229.6357.68實施例364.9486.81實施例4589.41336實施例561.78111.4實施例625.1652.34實施例7176.71672實施例8474.92289
[0157]
實驗結論:
[0158]
本發明化合物對穩定表達人源5-ht
2a
型hek393顯示了優異的細胞抗增殖活性。
[0159]
試驗例2人d2l受體功能試驗(激動劑檢測)
[0160]
細胞:穩定表達人源d
2l
的hek293細胞。
[0161]
試劑:dmem(invitrogen);pbs(corning);g418(invitrogen);blasticidin(invitrogen); penicillin/streptomycin(hyclone);fluo-4(invitrogen)。
[0162]
設備:384 well plate,greiner;vi-cell xr cell viability analyzer,beckman coulter;孵育箱, thermo。
[0163]
方法:將hek293細胞以20000/孔密度接種於384孔板,隔天測試化合物使用dmso配置,每孔加入0.75μl,測試濃度為10000nm、2500nm、625nm、156nm、39nm、9.76nm、 2.44nm、0.61nm、0.15nm、0.038nm,然後加入20μl fluo-4 directtm no-wash loading buffer 後放回孵箱,37℃,孵育50min,然後室溫孵育10min。使用flipr tetra高通量實時螢光檢測分析系統測定螢光值。化合物ec
50
、ec
90
使用prism計算得到,結果見下表。
[0164]
化合物編號ec
50
(nm)ec
90
(nm)
普拉克索4.3623.26實施例131.5》10000實施例211.2100.8實施例325.98236.9實施例4154685.8實施例542.33250.6實施例62152231實施例7377.52201實施例866.93732
[0165]
實驗結論:
[0166]
本發明化合物對穩定表達人源d
2l
型hek393細胞有良好的激動作用。
[0167]
試驗例3人d2l受體親和力試驗
[0168]
細胞:穩定表達人源d
2l
的hek293細胞。
[0169]
試劑:trisbase(sigma);polyethyleneimine(sigma);microscint20cocktail(perkinelmer);[3h]-7-oh-dapt(perkinelmer)。
[0170]
設備:unifilter-96gf/cfilterplates(perkinelmer);96wellconicalpolypropyleneplates(agilent);cellharvestc961961(perkinelmer);microbeta2reader(perkinelmer)。
[0171]
方法:測試化合物使用dmso配置,每孔加入1μl,測試濃度為100nm、25nm、6.25nm、1.56nm、0.39nm、97pm、24pm、6.1pm或10000nm、2500nm、625nm、156nm、39nm、9.76nm、2.44nm、0.61nm,然後加入100μl細胞膜和100μl同位素後密封,室溫下搖床300rpm孵育1h,同時用0.3%的polyethyleneimine浸泡unifilter-96gf/c過濾板。孵育完畢後用細胞收集器收集到gf/c過濾板,用洗板緩衝液洗4次後在50℃烘箱乾燥1h。將乾燥的gf/c過濾板底部封膜,每孔加入50μl閃爍液,並密封。使用microbeta2讀數。化合物ec
50
、ec
90
使用prism計算得到,結果見下表。
[0172]
化合物編號ec
50
(nm)ec
90
(nm)普拉克索5.6941..04實施例1540.37實施例20.727.08實施例30.827.37實施例42.7927.48實施例50.342.83實施例875.54-[0173]
實驗結論:
[0174]
本發明化合物對人源d2l受體具有較好的激動活性。
[0175]
試驗例4人5-ht2a受體親和力試驗
[0176]
細胞:穩定表達人源5-ht
2a
的hek293細胞。
[0177]
試劑:trisbase(sigma);polyethyleneimine(sigma);microscint20cocktail(perkinelmer);[3h]-ketanserin(perkinelmer)。
[0178]
設備:unifilter-96 gf/c filter plates(perkin elmer);96 well conical polypropylene plates (agilent);cell harvest c961961(perkin elmer);microbeta2 reader(perkin elmer)。
[0179]
方法:測試化合物使用dmso配置,每孔加入1μl,測試濃度為100nm、25nm、6.25 nm、1.56nm、0.39nm、97pm、24pm、6.1pm或10000nm、2500nm、625nm、156nm、 39nm、9.76nm、2.44nm、0.61nm,然後加入100μl細胞膜和100μl同位素後密封,室溫下搖床300rpm孵育1h,同時用0.3%的poly ethyleneimine浸泡unifilter-96 gf/c過濾板。孵育完畢後用細胞收集器收集到gf/c過濾板,用洗板緩衝液洗4次後在50℃烘箱乾燥1h。將乾燥的gf/c過濾板底部封膜,每孔加入50μl閃爍液,並密封。使用microbeta2讀數。化合物ic
50
、ic
90
使用prism計算得到,結果見下表。
[0180]
化合物編號ic
50
(nm)ic
90
(nm)匹莫範色林0.110.9實施例10.282.33實施例20.898.02實施例31.089.77實施例473.51635實施例52.4824.07實施例8》100-[0181]
實驗結論:
[0182]
本發明化合物對人源5-ht
2a
受體具有良好的抑制作用。
[0183]
藥理試驗表明,本發明提供的四氫苯並噻唑是一類全新的d
2l
和5-ht
2a
雙靶點調節劑,同時具有d
2l
受體的激動活性和5-ht
2a
受體的拮抗作用,相比於現有的單一作用的藥物具有重要的實用意義,且其激動活性和拮抗效果優於或接近現有的普拉克索、匹莫範色林等藥物的作用,可以用於製備治療/預防d
2l
和5-ht
2a
受體相關疾病的藥物的應用,例如治療帕金森病的運動症狀的同時預防/改善精神症狀、精神分裂症、精神病、分裂情感性障礙、雙相性精神障礙、高血壓繼發的精神病、偏頭痛、高血壓、血栓形成、血管痙攣、局部缺血、運動性抽搐、抑鬱、重度抑鬱症、焦慮、睡眠紊亂和食慾紊亂、帕金森病引起的非運動症狀、妄想、幻覺、抑鬱、焦慮、認知障礙、睡眠障礙、痴呆相關的精神疾病、精神分裂的陰性症狀、亨廷頓舞蹈病、阿爾茲海默症、脊髓小腦萎縮症、圖雷特氏症候群、馬查多-約瑟夫病、路易替痴呆、運動障礙、肌陣攣、震顫、進行性核上病麻痺,或其它對本領域技術人員而言顯而易見的其他疾病狀態和狀況等。
[0184]
以上所述僅為本發明的較佳實施例,並不用以限制本發明,凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護範圍之內。