一種具備雙單鏈末端PCR產物的獲得方法及其檢測方法與流程
2023-11-05 00:03:12 4
本發明屬於核酸技術領域,具體涉及一種具備雙單鏈末端pcr產物的獲得方法及其檢測方法。
背景技術:
聚合酶鏈式反應(pcr)是一種用於擴增特定的dna片段的分子生物學技術,它可看作是生物體外的特殊dna複製。pcr技術通過將模板核酸在高溫(95℃)時變性成單鏈,在降溫後(降至50℃-60℃)引物與模板根據鹼基互補配對特異性結合;在最適延伸反應溫度(72℃左右),dna聚合酶以5'-3'的方向合成互補鏈的反應。整個反應由高溫變性、低溫退火及適溫延伸組成的循環反應進行。一個循環需2~4分鐘,2~3小時使目的dna得以迅速擴增,將待擴目的基因擴增放大幾百萬倍。因pcr技術特異性強、靈敏度高、簡便、快速等優點而被廣泛應用。不僅常被用於核酸的基礎研究,pcr技術也是目前核酸檢測的金標準,在醫學、生物工程等領域應用廣泛。pcr產物是具有平末端的雙鏈dna,一般包括通過瓊脂糖凝膠電泳、聚丙烯醯胺凝膠電泳等進行分離,隨後需要藉助染色和紫外透射成像進行分析。上述操作步驟繁瑣、耗時長,且依賴儀器及特殊訓練的專業人員,不適用於高通量、快速的pcr產物檢測。
利用核酸試紙條進行檢測的技術是一項檢測pcr產物的新技術,該技術一般需要對pcr引物序列進行雙修飾或者其他特殊的處理:一、雙修飾引物:為了在試紙條上進行夾心法檢測都需要進行雙修飾常用於標記的小分子有biotin,digoxin,fitc,cy3等,。這些小分子都是修飾在引物的末端,當引物與模板退火延伸時,小分子也被摻入到在產物末端,從而達到標記pcr產物的目的。例如wansika等在檢測黑斑症候群和白斑症候群病毒核酸時,在兩條引物上分別標記biotin和fitc,因此鏈黴親和素(sa)包被的膠體金可以通過sa與biotin的高親和力作用而捕獲帶biotin-tag的擴增產物。由於在試紙條的t線區域事先劃有fitc的抗體,通過fitc抗體與抗原的特異性作用而將帶有fitc-tag的產物固定在t線區域,實現了對病毒核酸的快速檢測。這種修飾雖然可以實現快速檢測,但是需要對引物進行雙標記,增加了檢測的成本。二、雙修飾加高溫變性處理:pcr擴增產物一般都是平末端雙鏈,如果不在末端進行分子修飾的話,則沒有突出的粘性末端用於雜交;高溫變性可以將雙鏈分離而得到單鏈,通過與被標記在納米粒子表面的核酸互補配對而固定到納米粒子表面。例如despina等利用彩色聚苯乙烯納米微球標記上polyt作為tag,用於捕獲帶polya的目的核酸序列,用於快速靈敏地檢測玉米ivr基因(圖1)。ivr的pcr雙鏈片段高溫變性成單鏈後,與帶有polya的短鏈核酸雜交,形成帶polya尾巴的雙鏈片段,最後加入polyt聚苯乙烯納米微球將其捕獲;由於事先在pcr的primer上修飾了biotin(生物素),通過biotin和t線區域的sa高親和力形成納米微球-pcr產物-sa複合物,在t線區域形成可視化條帶。這樣通過雙重化學修飾再加上高溫變性才能用試紙條進行夾心法檢測,成本高且需要高溫變性來產生單鏈,效率低,影響檢測靈敏度。
現有的核酸檢測試紙條需對引物末端進行修飾才可以完成檢測,如專利cn105301237a公開了一種檢測核酸的膠體金標檢測試劑盒,就需對檢測線、質控線及探針使用抗原或半抗原,如生物素、地高辛或螢光染料等進行標記。新型的膠體金核酸試紙條是以膠體金作為示蹤標誌物,以鹼基互補配對原則為基礎的檢測材料,可以用於核酸的檢測。由於在t線、c線區固定是核酸,可以利用鹼基互補配對實現對目標序列的檢測。設計一段與待測產物(如,單鏈序列)一側單鏈互補的序列用於標金,t線上劃有與該產物另一側單鏈互補的序列,這樣當反應產物到達金標墊時通過鹼基互補配對作用將標金序列捕獲,形成產物-膠體金的複合物。在層析力的作用下此複合物沿著試紙條向前移動,由於t線序列能跟產物的另一側單鏈完全互補而將複合物進行固定,利用膠體金在t線處的聚集而形成肉眼可見的紅色條帶。c線上劃有與標金序列完全互補的序列,當多餘的膠體金到達c線時被捕獲而顯紅色,因此作為試紙條可以正常檢測的質控標準。由於常規pcr產物僅具有平末端結構,無法滿足單鏈雜交的條件,該方法尚無法進行pcr產物的檢測。
紫外線指的是電磁波譜中波長從100nm~400nm輻射的總稱,可分為uva(波長320~400nm)、uvb(波長290~320nm)、uvc(波長100~290nm)。已有研究表明,紫外輻射會給生物體帶來有害影響,包括基因誘變、致癌作用及細胞死亡等,產生這些有害影響的可能原因是在dna分子內生成了光化學反應產物。在uv的作用下,核酸序列相鄰的嘧啶鹼基經[2+2]環加成反應形成環丁烷二聚體,其中胸腺嘧啶二聚體是主要的光致產物。。
技術實現要素:
針對平末端pcr產物造成的核酸檢測方法繁瑣複雜、成本高、靈敏度低的問題,本發明提出一種新的解決方案,無需特殊修飾及其它操作步驟,就可以直接對pcr產物進行檢測,達到準確、快速、靈敏、低成本、操作簡易的檢測需求。
本發明的一個目的是提供一種具備雙單鏈末端pcr產物的獲得方法,包括如下步驟:
(1)設計用於擴增目的核酸的正向引物f和反向引物r序列,並在正向引物f和反向引物r序列的5'端分別加上tag1和tag2序列,tag1序列和正向引物f序列之間、tag2序列和反向引物r序列之間分別用若干胸腺嘧啶隔開,合成引物tag1-f和tag2-r;
(2)用紫外線對引物tag1-f和tag2-r的水溶液照射至少30min;
(3)混合pcr反應體系置於pcr儀中進行反應,得到具備雙單鏈末端的pcr產物。
進一步地,步驟(1)中若干胸腺嘧啶的兩端分別各加一個腺嘌呤或胞嘧啶,可進一步提高二聚體的形成率。由於tt-cpd(胸腺嘧啶二聚體)吸收光子變成激發態後,很有可能被鄰近鹼基猝滅,這可能依賴於他們的遷移能或電荷重疊的幾何中心,比如鹼基和鹼基的重疊決定了電子耦合,因此鄰近鹼基通過影響二聚體的回覆並影響其最終生成率。特別是當鄰近鹼基為鳥嘌呤(g)的時候,回復率最高,導致形成率最低,而使用腺嘌呤或胞嘧啶時,形成率則較高。
進一步地,步驟(1)中胸腺嘧啶的數量為6-10個。
進一步地,步驟(1)中正向引物和反向引物序列長度分別為18-25nt,tag1和tag2序列長度都為12nt。根據不同待檢基因可以調整正向引物和反向引物序列,tag1和tag2序列可以通用。tag1和tag2序列可根據引物設計原則進行隨機設計,只要不含連續的t即可。
進一步地,tag1和tag2序列為:
tag1:5'-ctgtacgagact-3'
tag2:5'-tctaatgctgat-3',
上述tag1和tag2序列已證實可以在核酸檢測中通用。
進一步地,步驟(2)中紫外線優選波長100~290nm。
本發明的另一個目的是提供上述具備雙單鏈末端pcr產物的檢測方法,可選擇以下兩種方法中的任一種:
(一)直接法:分別設計併合成與tag1和tag2互補的序列c-tag1和c-tag2,並分別在c-tag1和c-tag2的3'端用巰基(-sh)修飾;將修飾過後的c-tag1和c-tag2均通過金硫鍵(au-s)結合到膠體金粒子上,得到兩種不同的膠體金標記產物:aunpdna1和aunpdna2;將aunpdna1和aunpdna2按c-tag1和c-tag2摩爾比0.8-1.2:0.8-1.2混勻製備成紅色透明的標記膠體金溶液;將pcr產物加入到標記膠體金溶液中混勻,肉眼觀察進行檢測及判斷,標記膠體金溶液無變化則檢測結果為陰性,標記膠體金溶液變渾濁則檢測結果為陽性;
(二)試紙條法:分別設計併合成與tag1和tag2互補的序列c-tag1和c-tag2,c-tag1的3'端用巰基(-sh)修飾,合成tag1序列;將修飾後的c-tag1通過金硫鍵(au-s)結合到膠體金粒子上,得到aunpdna1,並將aunpdna1噴在膠體金試紙條的金標墊上;將c-tag2序列固定在試紙條的檢測區(t);將tag1序列固定在試紙條的質控區(c);將pcr產物滴加到試紙條的樣品墊,滴加上樣緩衝液,等待觀察結果。
方法(一)利用膠體金的等離子體共振效應,分散的膠體金顆粒呈紅色而聚集的膠體金顆粒呈紫色、藍色,並出現沉澱。因此,通過dna的雜交可將表面具有dna標記的膠體金連接在一起,「拉近」膠體金顆粒之間的距離,並組裝成為大分子顆粒從而產生聚沉,此時溶液出現紅移現象,顏色改變,並出現肉眼可見的沉澱顆粒,因此可實現對pcr產物的快速檢測。本方法的原理如圖6所示,形成兩側均有單鏈末端的pcr產物後,在膠體金的表面通過au-s配位鍵修飾了兩種不同單鏈dna,它們分別可以與pcr產物末端的tag1和tag2雜交,並發生上述變化。
方法(二)利用pcr產物單鏈末端tag1與試紙條金標墊上aunpdna1互補,tag2與試紙條檢測區上c-tag2互補,通過這兩條tag的夾心作用,pcr產物可以被捕獲並固定在檢測區處產生紅線,缺少一條tag都不可能出現紅色t線;質控區固定的核酸tag1可以通過與aunpdna1互補而捕獲多餘的膠體金呈現紅色。因此陽性為兩條紅線,陰性為一條紅線(質控區)。
進一步地,方法(二)中採用劃膜噴金儀將aunpdna1、c-tag2序列、tag1序列噴或劃在膠體金試紙條上。
進一步地,方法(二)中上樣緩衝液為:4×ssc,ph7.0,滴加量為10-50μl。
進一步地,方法(二)中pcr產物的上樣量為5-10μl。
本發明的另一個目的是提供一種核酸檢測試紙條,其依次包括樣品墊、金標墊、硝酸纖維素膜和吸水墊模塊,每相鄰的兩模塊部分重疊,硝酸纖維素膜上依次設置有檢測區和質控區;其pcr的正向引物和反向引物序列的5'端分別加上tag1和tag2序列,tag1序列和正向引物序列之間、tag2序列和反向引物序列之間分別用若干胸腺嘧啶隔開;將tag1的互補序列c-tag1與膠體金製備成核酸-金標記物aunpdna1,並將aunpdna1噴在金標墊上;將與tag2互補的序列c-tag2固定在檢測區;將tag1序列固定在質控區。
進一步地,樣品墊、金標墊、硝酸纖維素膜和吸水墊都固定在pvc板上。
本發明的另一個目的是提供上述核酸檢測試紙條的製備方法,包括如下步驟:(1)將製備好的核酸-金標記物aunpdna1用劃膜噴金儀噴在玻璃纖維膜上製成金標墊;(2)將c-tag2序列和tag1序列分別用劃膜噴金儀劃在硝酸纖維素膜上,分別構成檢測區和質控區;(3)將樣品墊、金標墊、硝酸纖維素膜和吸水墊模塊分別組裝在pvc板上,每相鄰的兩模塊重疊1-3mm;(4)將組裝好的試紙條切割成2-5mm寬。
本發明的另一個目的是提供一種核酸檢測試劑盒,包括試紙條體系和/或標記膠體金溶液;
所述試紙條體系包括核酸檢測試紙條和上樣緩衝液;
所述核酸檢測試紙條依次包括樣品墊、金標墊、硝酸纖維素膜和吸水墊模塊,每相鄰的兩模塊部分重疊,硝酸纖維素膜上依次設置有檢測區和質控區;其pcr的正向引物f和反向引物r序列的5'端分別加上tag1和tag2序列,tag1序列和正向引物f序列之間、tag2序列和反向引物r序列之間分別用若干胸腺嘧啶隔開;將tag1的互補序列c-tag1與膠體金製備成核酸-金標記物aunpdna1,並將aunpdna1噴在金標墊上;將與tag2互補的序列c-tag2固定在檢測區;將tag1序列固定在質控區;所述樣品墊、金標墊、硝酸纖維素膜和吸水墊都固定在pvc板上;
所述上樣緩衝液為:4×ssc,ph7.0;
所述標記膠體金溶液由aunpdna1和aunpdna2按c-tag1和c-tag2摩爾比0.8-1.2:0.8-1.2混和而成;所述aunpdna1和aunpdna2分別為表面修飾c-tag1和c-tag2的膠體金粒子。
本申請人利用紫外照射在引物中形成胸腺嘧啶二聚體,發現可以阻隔pcr聚合酶的延伸作用,使用這種帶有胸腺嘧啶二聚體的引物對目的片段進行pcr擴增,可以使pcr產物末端帶有單鏈序列,因此可以使用膠體金核酸試紙條對其進行檢測。本發明通過結合含有胸腺嘧啶二聚體引物的pcr技術和核酸試紙條技術,建立了一種簡便快捷的檢測方法,從而省去電泳等步驟,達到高效、準確、快速、靈敏、低成本、操作簡易的檢測需求。該方法具有以下優勢:
(1)與普通的核酸檢測試紙條相比,本發明用pcr這一核酸檢測的金標準作為信號放大手段,大大提高了檢測靈敏度和通用性。
(2)與現有的阻隔pcr反應不同,本發明不需要對引物進行特殊的修飾,只需要簡單的紫外照射處理,就可以達到形成單鏈末端pcr產物的目的,處理過程簡單、無需繁瑣的修飾步驟、成本低廉。
(3)與傳統的引物修飾的方法相比,本發明省去了修飾的步驟,紫外照射完成後就可以進行pcr反應,不受人員、地區等條件的限制,尤其適用於現場實時檢測或缺乏儀器的偏遠地區使用。若pcr擴增產物即可直接用標記膠體金溶液或試紙條檢測,無需用到抗體或者配體等蛋白,節約了檢測成本。
(4)本發明中的tag1和tag2序列可以通用,只要更換與待測目標互補的引物序列,檢測所用標記膠體金溶液或試紙條都不用做調整,可直接使用,是一種廣譜檢測平臺,值得推廣使用。
(5)使用方便快速,便於實時檢測和偏遠地區使用,反應在10分鐘內即可完成;成本低,不需要特殊的儀器設備;應用範圍廣,可適應多種檢測對象;膠體金本身為紅色,不需要加入發色試劑,便於觀察。
附圖說明
圖1現有技術中聚苯乙烯納米微球檢測ivr的pcr產物示意圖;
圖2實施例1變性膠電泳結果;
圖3實施例2含有胸腺嘧啶的引物示意圖;
圖4胸腺嘧啶二聚體生成機制;
圖5實施例2中pcr反應過程;
圖6實施例3中利用標記膠體金溶液檢測pcr產物的原理圖;
圖7實施例3中膠體金溶液變化結果;
圖8實施例4試紙條結構示意圖;
圖9實施例4中核酸試紙條檢測pcr產物結果及原理;
圖10實施例4中pcr產物電泳結果。
具體實施方式
下面結合具體實施例及附圖對本發明做進一步詳細說明。
實施例1胸腺嘧啶二聚體阻隔pcr聚合酶延伸
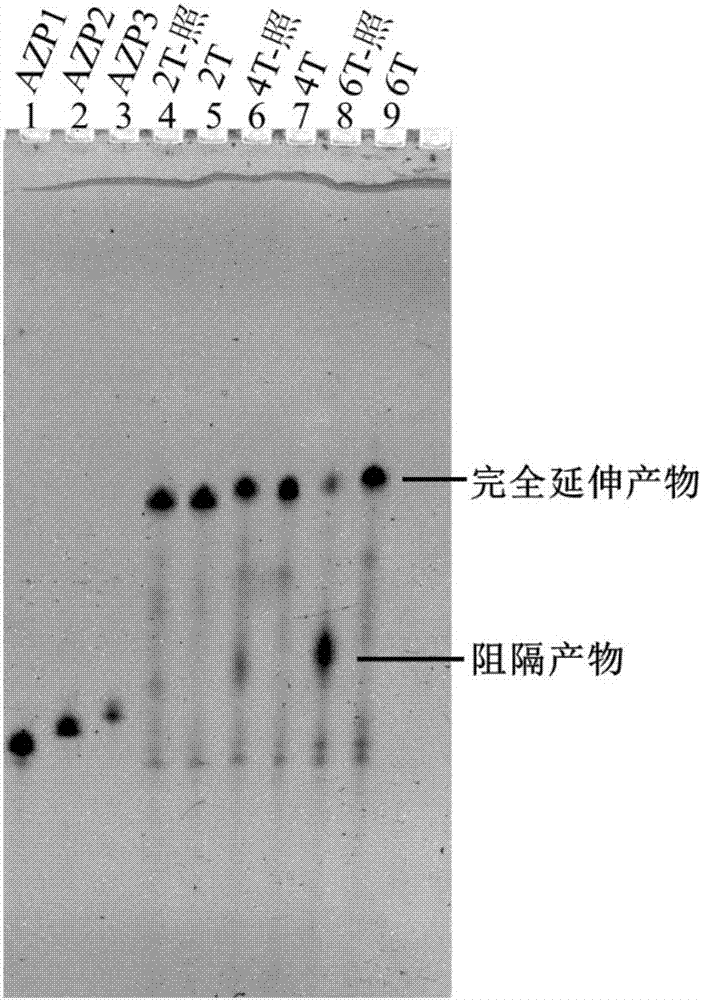
以含有胸腺嘧啶的核酸序列為模板,2t、4t、6t分別表示模板中含有2、4、6個相鄰的胸腺嘧啶(如表1所示),用azp1作為引物,進行引物延伸實驗,即只進行單個循環(35℃退火5min,72℃延伸5min)。實驗結果如圖2所示:用變性page膠進行產物分析,在模板沒有經過紫外線照射的情況下,azp1能以2t、4t、6t為模板完全延伸(泳道5、7、9);當用100~290nm波長紫外線照射模板40min後,以4t和6t為模板的情況下(泳道6、8),azp1延伸了若干鹼基之後就不再延伸,有明顯的阻隔產物形成,以6t為模板的情況下最為明顯。azp1大約延伸了5-6個鹼基,條帶位置後移約5個鹼基停在24nt的位置。
表1實施例1所涉及序列信息
由於模板中含有胸腺嘧啶二聚體,pcr聚合酶在胸腺嘧啶作用下鹼基延伸被阻隔而無法形成完全互補的雙鏈,因此將這種含有胸腺嘧啶二聚體的引物用於pcr反應,可產生兩側都有單鏈的pcr產物。從上面的實驗可以證明紫外照射形成的胸腺嘧啶二聚體阻隔了pcr聚合酶的延伸。
實施例2伊波拉病毒部分序列的pcr擴增
(1)根據genbank中已發布的伊波拉病毒基因組序列選定待pcr擴增的dna片段,序列信息見表2,總長103nt。分別設計正向引物f和反向引物r,並在f、r的5'端分別加上tag1和tag2序列,f和tag1序列之間、r和tag2序列之間分別用6個胸腺嘧啶隔開,分別得到tag1-f和tag2-r。引物及模板關係見圖3。
表2ebolatarget單鏈末端pcr產物擴增實驗所需序列信息
(2)用uvc(波長100~290nm)對實驗組用的tag1-f和tag2-r的水溶液進行照射30min,使相鄰的胸腺嘧啶形成二聚體,分子機制見圖4;未照射對照組的引物不經uvc照射。
(3)混合pcr反應體系置於pcr儀中進行反應:設置不加模板ebolatarget的空白對照組,實驗組及未照射對照組除用的引物分別為照射和未照射過的外,其餘條件均相同。pcr反應體系及pcr循環參數分別見表3、表4;pcr反應過程如圖5所示。
表3pcr實驗反應體系:
表4pcr實驗反應條件:
step2-4循環30次。
實施例3pcr擴增產物的直接法檢測
(1)利用檸檬酸鈉還原法製備膠體金顆粒:在250ml錐形瓶中加入96ml超純水,用移液器移取1ml1wt%的haucl4溶液加入其中,同時加入攪拌磁子,攪拌的同時,加熱至沸騰。隨後,迅速加入3ml1wt%的檸檬酸鈉溶液,繼續攪拌並保持微沸。溶液顏色隨即變黑,繼續加熱攪拌,可觀察到溶液顏色由深變淺,最後由磚紅色變為酒紅色。等溶液顏色穩定不再變化後,繼續加熱攪拌10min。繼續攪拌,待溶液降至室溫,製備得到粒徑15±2nm的膠體金顆粒。
(2)在膠體金顆粒表面通過au-s配位鍵標記兩種不同單鏈dna(即為標金序列:分別為c-tag1、c-tag2(表2),其3'端進行巰基修飾)。在2ml膠體金溶液中加入50μl標金序列(10μmol/l,c-tag1/c-tag2),225μl水,350μl100mmol/lpbs(ph8.0),350μl0.1wt%sds,525μl2mol/lnacl(視膠體金溶液穩定性差異進行分步滴加,逐次提高標金溶液中的nacl濃度),室溫放置過夜。12000r/min離心10min,棄上清並用1ml0.01wt%sds洗滌沉澱,重複操作兩次,沉澱重溶於1ml保存液(0.3mol/lnacl、10mmol/lpbs、0.01%sds)中。至此,製備得到兩種表面修飾有單鏈dna的膠體金溶液。
(3)利用表面修飾dna的膠體金對pcr產物進行檢測,原理如圖6所示。取20μl實施例2的pcr擴增產物,加入40μlc-tag1修飾的膠體金溶液和40μlc-tag2修飾的膠體金溶液,加入1/4體積分數的乙醇後混勻靜置,經10-20min即可觀察到可視溶液變化出現。如圖7所示的結果,未加入pcr產物、空白對照組及未照射對照組的膠體金溶液顏色均沒有改變,而加入pcr產物後的膠體金溶液顏色發生變化,說明pcr產物可以在膠體金溶液中充當linker的作用,能夠拉近膠體金之間的距離,並組裝形成大分子顆粒,最終使膠體金顆粒發生聚沉,通過肉眼可以觀察到溶液的顏色變化及沉澱出現。該方法證明了經過uv照射的引物能夠使pcr產物末端形成兩條單鏈,且用該方法可直接針對pcr產物進行檢測。
實施例4pcr擴增產物的試紙條法檢測
(1)分別設計併合成與tag1和tag2互補的序列c-tag1和c-tag2,c-tag1的5'端用巰基(-sh)修飾,合成tag1序列。
(2)將修飾後的c-tag1通過金硫鍵(au-s)結合到膠體金粒子上,得到核酸-金標記物aunpdna1,並將aunpdna1噴在膠體金試紙條的金標墊(玻璃纖維膜)上;將c-tag2序列用劃膜噴金儀固定在硝酸纖維素膜的檢測區(t);將tag1序列用劃膜噴金儀固定在硝酸纖維素膜的質控區(c)。
如圖8所示,將樣品墊、金標墊、硝酸纖維素膜和吸水墊模塊分別組裝在pvc板上,每相鄰的兩模塊重疊2mm,保證層析過程順利進行;將組裝好的試紙條用自動斬切機,切割成3mm寬。
(3)將5μl實施例2的pcr產物滴加到試紙條的樣品墊(實驗組和對照組分別滴加到不同試紙條),逐滴滴加15μl4×ssc,讓樣品在試紙條上緩慢前行,5分鐘後觀察結果,實驗組在檢測區(t)和質控區(c)各出現一條紅色條帶,說明pcr反應正常進行並得到了目標產物,檢測結果及原理見圖9。未照射對照組和空白對照組在檢測區(t)和質控區(c)均未出現條帶。
另外,為驗證試紙條的可靠性,對上述pcr產物同時進行了傳統的電泳檢測,如圖10所示,圖中可見,未照射對照組在150bp左右有特異性擴增條帶,實驗組在200bp左右有目標產物,空白對照組無特異性條帶。充分證實上述直接法和試紙條法檢測pcr產物的準確度高。
相比於傳統的電泳檢測pcr產物的方法,本發明的方法無需進行引物修飾,pcr產物可直接進行檢測,並節省了電泳、染色的過程,檢測更簡單、省時,準確度高、靈敏度高,pcr產物檢測用量可微至5μl,僅需5-10min即可得到檢測結果。
sequencelisting
中國海洋大學
一種具備雙單鏈末端pcr產物的獲得方法及其檢測方法
2017
13
patentinversion3.5
1
36
dna
artificialsequence
2t
1
tctaatgctgatgttacatcgtcgtcgtccaaattg36
2
38
dna
artificialsequence
4t
2
tctaatgctgatgttttacatcgtcgtcgtccaaattg38
3
40
dna
artificialsequence
6t
3
tctaatgctgatgttttttacatcgtcgtcgtccaaattg40
4
19
dna
artificialsequence
azp1
4
caatttggacgacgacgat19
5
103
dna
artificialsequence
ebolatarget
5
gttgacccgtatgatgatgagagtaataattatcctgactacgaggattcggctgaaggc60
accacaggagatcttgatctcttcaatttggacgacgacgatg103
6
24
dna
artificialsequence
f
6
gttgacccgtatgatgatgagagt24
7
20
dna
artificialsequence
r
7
catcgtcgtcgtccaaattg20
8
12
dna
artificialsequence
tag1
8
ctgtacgagact12
9
12
dna
artificialsequence
tag2
9
tctaatgctgat12
10
44
dna
artificialsequence
tag1-f
10
ctgtacgagactattttttagttgacccgtatgatgatgagagt44
11
40
dna
artificialsequence
tag2-r
11
tctaatgctgatattttttacatcgtcgtcgtccaaattg40
12
12
dna
artificialsequence
c-tag1
12
agtctcgtacag12
13
12
dna
artificialsequence
c-tag2
13
atcagcattaga12