一種PS/cPGMA核殼型納米粒及其製備方法與流程
2023-11-05 04:12:42 2
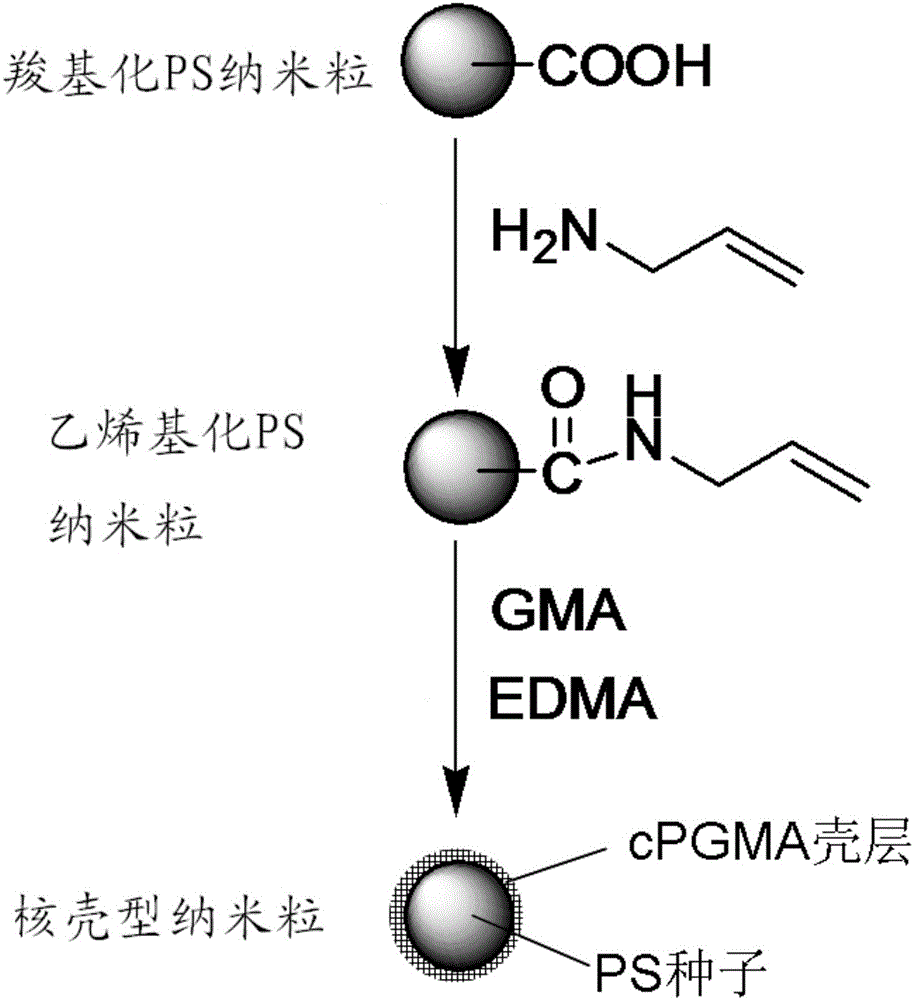
本發明屬於親和高分子微球製備領域,具體涉及一種PS/cPGMA核殼型納米粒及其製備方法。
背景技術:
膠乳增強免疫比濁法(LETIA),通過採用物理吸附或共價鍵合的方式將抗體(或抗原)偶聯到納米顆粒的表面,形成納米粒-抗體(抗原)複合物,此複合物與樣品中的抗原(或抗體),通過抗體抗原反應,形成納米粒-抗體-抗原聚集顆粒,隨著免疫反應的不斷發生,聚集的顆粒不斷增大,從而導致溶液在一定波長(如600nm)的吸光值發生顯著的變化,通過測定免疫反應前後吸光度值的變化可以計算出樣品中抗原(抗體)的濃度,從而達到檢測的目的。同其它免疫分析方法(如:放射免疫分析法、化學發光免疫分析法、酶聯免疫分析法等)相比,LETIA由於具有檢測方法簡單方便、無放射性汙染、可準確定量、穩定性好、同時可在全自動生化儀上實現高通量樣品檢測等優點,已經越來越多的被應用於臨床檢測。迄今,LETIA在臨床中已經應用於檢測β2-微球蛋白、胱抑素C、D-二聚體、脂蛋白a、抗鏈球菌溶血素「O」、類風溼因子、C-反應蛋白(CRP)、高敏C反應蛋白(hsCRP)、α1-微球蛋白、肌鈣蛋白、肌紅蛋白、肌酸激酶同工酶、甲胎蛋白、胃蛋白酶原I、前列腺特異性抗原、糖化血紅蛋白等多種檢測項目,診斷領域已涉及腎功能、風溼、心肌、腫瘤、糖尿病等多個領域,擁有廣闊的市場前景。
LETIA中通常使用聚苯乙烯(PS)材料的納米級膠乳顆粒,抗體可以通過與PS的疏水性相互作用以物理吸附的形式固定於納米粒表面,然而這種方法製備的抗體-納米粒複合物穩定性較差。一般通過對PS納米粒進行表面改性或共聚等方法在納米粒表面引入活性基團,進而採用化學鍵和的 方法進行抗體固定化可有效提高試劑的穩定性。
通過與丙烯酸共聚得到的羧基化PS納米粒在LETIA中最常採用,例如Kyhse-Andersen等(J.Kyhse-Anderson,C.Schmidt.G.Nordin,B.Andersson,et.al.,Serum cystatin C,determined by a rapid,automated particle-enhanced turbidimetric method,is a better marker than serum creatinine for glomerular filtration rate.Clin.Chem.,1994,40(10),1921-1926.)採用羧基化PS納米粒製備了光抑素C抗體固定化的PS納米顆粒,並建立了血漿光抑素C的LETIA檢測方法。採用化學鍵合的方法在PS納米粒表面實現蛋白固定化顯著提高了試劑的穩定性,但是由於固定化基質材料(如羧基化PS納米粒)表面仍有苯環基團暴漏,固定在納米粒表面的蛋白質仍將有一部分物理吸附(而非化學鍵合)在納米粒表面。例如Santos和Forcada(R.M.Santos,J.Forcada.Acetal-functionalized polymer particles useful forimmunoassays.Ⅲ:preparation of latex-protein complexes and their applications.Journal of Materials Science:Materials in Medicine.2001,12:173-180.)報導採用表面醛基修飾的PS納米粒進行抗體固定化,其中仍有約20%的抗體以物理吸附的形式固定化。
為了進一步的避免蛋白質與膠乳納米粒的物理吸附作用,使蛋白質完全以化學鍵合的形式固定到膠乳納米粒表面,並在此基礎上增加蛋白質-納米粒複合物的穩定性,需對納米粒的表面進行徹底的親水性改造,即納米粒表面覆蓋一層親水性塗層使納米粒對蛋白質無非特異性吸附作用。早在1984年Litchfield(W.J.Litchfield,A.R.Craig,W.A.Frey,C.C.Leflar,C.E.Looney,M.A.Luddy.Novel shell/core particles for automated turbidimetric immunoassays.Clinical Chemistry,1984,30:1489-1493.)等便利用種子乳液聚合法,製備了外殼親水的「核殼型」納米顆粒。該方法中採用PS納米粒為種子,甲基丙烯酸縮水甘油酯(GMA)為外殼聚合單體,製備了以PS為種子,聚GMA(PGMA)為塗層材料的PS/PGMA「核殼型」納米粒子,並將其用於製備LETIA檢測試劑。採用類似的方法,段殷等(段殷,左鋒,柴 宏森,劉建華,王冬梅,徐亮,周晶。單分散PS/PGMA核-殼型高分子微球的製備與表徵。天津醫科大學學報,2012,18:416-418.)也製備了PS/PGMA納米粒,並系統的考察了種子納米粒的粒徑、GMA的加入量、乳化劑的加入量、聚合反應時間等因素對核-殼型納米粒製備的影響。
上述方法雖然通過製備「核殼型」納米材料實現了納米粒表面的親水性改造,但是具有親水性的外殼聚合物材料是以物理吸附的方式吸附在PS種子納米粒表面,這易導致「核殼型」納米粒子的穩定性變差。此外,外殼塗層材料PGMA親水性較強,在水溶液中長期貯存易溶脹,導致製得的核殼型納米粒的水力學粒徑變大。
技術實現要素:
本發明的目的在於,為了解決現有技術中核殼型PS納米粒的外殼聚合物材料是以物理吸附的方式吸附在PS納米粒表面,從而存在穩定性差;並且在水溶液中長期貯存易溶脹,導致納米粒水力學粒徑變大的問題,提供一種PS/cPGMA核殼型納米粒的製備方法。
為了達到上述目的,本發明的技術方案如下:
一種PS/cPGMA核殼型納米粒的製備方法,包括步驟如下:
(1)將羧基化PS納米粒表面進行乙烯基化修飾得到乙烯基化PS納米粒;
(2)將乙烯基化PS納米粒通過種子乳液聚合法製備PS/cPGMA核殼型納米粒。
上述PS/cPGMA核殼型納米粒的製備方法中,所述羧基化PS納米粒的平均粒徑為60nm-500nm,羧基密度以幹球計為0.065mmol/g-0.350mmol/g。
上述PS/cPGMA核殼型納米粒的製備方法中,所述進行乙烯基化修飾的方法包括以下步驟:將羧基化PS納米粒分散,經1-(3-二甲氨基丙基)-3- 乙基碳二亞胺鹽酸鹽活化後,再加入丙烯胺進行反應,得到乙烯基化PS納米粒。
上述PS/cPGMA核殼型納米粒的製備方法中,加入的1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽的物質的量為羧基化PS納米粒上羧基的物質的量的1-5倍。
上述PS/cPGMA核殼型納米粒的製備方法中,將羧基化PS納米粒分散於磷酸鹽緩衝液中,得到PS納米粒的水溶液,其中羧基化PS納米粒的質量為磷酸鹽緩衝液質量的2-10%;所述丙烯胺以體積濃度為10%的丙烯胺磷酸鹽緩衝液的形式加入,加入量為PS納米粒水溶液體積的0.5-1.5;所述磷酸鹽緩衝液為pH7.4、25mM的磷酸鹽緩衝液;所述活化的條件為:室溫震蕩反應15-60分鐘;加入丙烯胺後於4-37℃震蕩,反應的時間為3-12小時。
上述PS/cPGMA核殼型納米粒的製備方法中,所述種子乳液聚合法製備PS/cPGMA核殼型納米粒的方法包括:
以乙烯基化PS納米粒為種子,過硫酸鉀為引發劑,GMA為單體,碳酸鹽緩衝液為分散相,十二烷基磺酸鈉為表面活性劑,乙二醇二甲基丙烯酸酯為交聯劑。
上述PS/cPGMA核殼型納米粒的製備方法中,所述種子乳液聚合法製備PS/cPGMA核殼型納米粒的具體方法包括:
將乙烯基化PS納米粒水溶液與碳酸鹽緩衝溶液按體積比為1:1-1:3混合;然後加入過硫酸鉀、GMA、乙二醇二甲基丙烯酸酯和十二烷基磺酸鈉攪拌反應,攪拌轉速為120轉每分鐘,反應溫度為70-80℃,反應時間為6-10h,製得PS/cPGMA納米粒;所述乙烯基化PS納米粒水溶液中的水溶液為pH7.4、25mM的磷酸鹽緩衝液,乙烯基化PS納米粒的質量為磷酸鹽緩衝液質量的2-10%。
上述PS/cPGMA核殼型納米粒的製備方法中,過硫酸鉀的質量與乙烯基化PS納米粒水溶液的體積比為0.1-0.5g/100mL,GMA的質量與乙烯基化PS納米粒水溶液的體積比為0.2-2g/100mL,十二烷基磺酸鈉的質量與乙烯基化PS納米粒水溶液的體積比為0.01-0.05g/100mL;乙二醇二甲基丙烯酸酯的質量為GMA質量的1-5%。
上述PS/cPGMA核殼型納米粒的製備方法製備的PS/cPGMA核殼型納米粒。
上述PS/cPGMA核殼型納米粒在膠乳增強免疫比濁法中的應用。
與現有技術相比,本發明的優點如下:
1、本發明的PS/cPGMA核殼型納米粒的製備方法,對羧基化PS納米粒表面進行乙烯基團活化,使殼層聚合物材料以化學鍵合的方式固定在羧基化PS種子納米粒表面,提高製得納米粒的穩定性。
2、本發明的PS/cPGMA核殼型納米粒的製備方法,在PGMA殼層材料的製備中,加入了交聯劑EDMA,EDMA的投料量佔GMA投料量的1-5%,在保證納米粒表面的親水性的同時,從而控制殼層材料PGMA在水溶液中的溶脹,進一步提高製得納米粒的穩定性。
3、本發明的PS/cPGMA核殼型納米粒的製備方法,選擇粒徑範圍在60nm-500nm的羧基化PS納米粒為原料,通過EDC活化後易與帶有氨基的丙烯胺反應,進而在納米粒表面引入乙烯基團,為後續種子乳液聚合法製備PS/cPGMA核殼型納米粒製得一定粒徑的乙烯基化PS種子納米粒。
4、本發明的PS/cPGMA核殼型納米粒的製備方法製備的PS/cPGMA核殼型納米粒外殼親水且在水溶液中穩定性好;外殼親水可確保蛋白質以化學鍵合的方式,而非物理吸附固定到納米粒表面,與蛋白質非特異性吸附作用較弱,可作為LETIA試劑的生產原料使用,與PS納米粒製備的LETIA試劑相比穩定性顯著提高。
附圖說明
下面將通過附圖和具體實例進一步說明本申請。
圖1為PS/cPGMA納米粒製備過程示意圖。
圖2為PS/cPGMA納米粒的透射電鏡照片。
圖3為光抑素C檢測試劑穩定性測試結果對比;其中,圖(A)為PS/cPGMA納米粒製備的試劑;圖(B)為PS納米粒製備的試劑。
圖4為本發明製備的PS/cPGMA核殼型納米粒與PS/PGMA納米粒的穩定性結果。
具體實施方式
本發明的所有實施例中的原料、試劑如無特殊說明均為市售商品。下面結合實例對本發明的實施進一步進行說明,但是本發明的實施不僅限於此。
實施例1
根據圖1所示的製備過程示意圖,製備平均粒徑約為120nm的PS/cPGMA納米粒;
將羧基化PS納米粒(瑞安市伊普西隆生物科技有限公司)分散於PBS(25mM,pH7.4)中,製備5%(w/w)的PS納米粒水溶液100mL。羧基化PS納米粒平均粒徑為109.6nm,羧基密度為0.157mM/g(幹球),加入150.5mg EDC,室溫震蕩反應15分鐘後,繼續加入100mL含10%(v/v)丙烯胺的PBS(25mM,pH7.4)溶液。25℃震蕩反應8小時後,用去離子水透析除去未反應的丙烯胺,製得乙烯基化PS納米粒。用截留分子量為300KD的超濾膜將製得的乙烯基化PS納米粒濃縮至100mL,5%(w/w)的水溶液。
將上述製備的100mL乙烯基化PS納米粒水溶液置於一圓底三口燒瓶中,分別加入200mL碳酸鹽緩衝液(pH9.0,25mM),0.2g KPS,0.8g GMA,0.016g EDMA,0.02g SDS,在充氮氣的條件下,置於75℃水浴中,120轉 每分鐘攪拌反應8h,製得PS/cPGMA納米粒。將製得的納米粒用去離子水透析,並超濾濃縮至5%(w/w)的水溶液。
將製得的PS/cPGMA納米粒用透射電子顯微鏡觀測,結果參見圖2。由圖可知,製得的微球具有較好的球形度,粒度分布均勻(圖2A),具有明顯的核殼型結構(圖2B)。通過粒徑分析儀測定,製得的PS/cPGMA納米粒的平均粒徑為120.6nm,比種子納米粒的粒徑略有增加(種子納米粒即羧基化PS納米粒的平均粒徑為109.6nm),粒徑增大的部分必然是由於納米粒殼層的存在而導致,這也與圖2顯微鏡觀測的結果一致。
實施例2
根據圖1所示的指標過程示意圖,製備平均粒徑約為65nm的PS/cPGMA納米粒;
將羧基化PS納米粒(瑞安市伊普西隆生物科技有限公司)分散於PBS(25mM,pH7.4)中,製備10%(w/w)的PS微球水溶液100mL。羧基化PS納米粒平均粒徑為60.0nm,羧基密度為0.065mM/g(幹球),加入124.6mg EDC,室溫震蕩反應15分鐘後,繼續加入50mL含10%(v/v)丙烯胺的PBS(25mM,pH7.4)溶液。4℃震蕩反應12小時後,用去離子水透析除去未反應的丙烯胺,製得乙烯基化PS納米粒。用截留分子量為300KD的超濾膜將製得的乙烯基化PS納米粒濃縮至100mL,10%(w/w)的水溶液。
將上述製備的100mL乙烯基化PS納米粒水溶液置於一圓底三口燒瓶中,分別加入300mL碳酸鹽緩衝液(pH9.0,25mM),0.5g KPS,2g GMA,0.02g EDMA,0.05g SDS,在充氮氣的條件下,置於70℃水浴中,120轉每分鐘攪拌反應10h,製得PS/cPGMA納米粒。將製得的納米粒用去離子水透析,並超濾濃縮至5%(w/w)的水溶液。用粒徑分析儀測定製得的PS/cGMA納米粒的平均粒徑為65.0nm。
實施例3
根據圖1所示的指標過程示意圖,製備平均粒徑約為520nm的PS/cPGMA納米粒;
將羧基化PS納米粒(瑞安市伊普西隆生物科技有限公司)分散於PBS(25mM,pH7.4)中,製備2%(w/w)的PS微球水溶液100mL。羧基化PS納米粒的平均粒徑為500nm,羧基密度為0.350mM/g(幹球),加入134.2mg EDC,室溫震蕩反應15分鐘後,繼續加入150mL含10%(v/v)丙烯胺的PBS(25mM,pH7.4)溶液。37℃震蕩反應3小時後,用去離子水透析除去未反應的丙烯胺,製得乙烯基化PS微球。用截留分子量為300KD的超濾膜將製得的乙烯基化PS納米粒濃縮至100mL,2%(w/w)的水溶液。
將上述製備的100mL乙烯基化PS納米粒水溶液置於一圓底三口燒瓶中,分別加入100mL碳酸鹽緩衝液(pH9.0,25mM),0.1g KPS,0.2g GMA,0.01g EDMA,0.01g SDS,在充氮氣的條件下,置於80℃水浴中,120轉每分鐘攪拌反應6h,製得PS/cPGMA納米粒。將製得的納米粒用去離子水透析,並超濾濃縮至5%(w/w)的水溶液。用粒徑分析儀測定製得的PS/cGMA納米粒的平均粒徑為520.1nm。
實施例4
PS/cPGMA納米粒製備的光抑素C(Cys-C)檢測試劑的穩定性考察。
取實施例1製備的PS/cPGMA微球水溶液(5%w/w)20mL,加入20mL碳酸鹽緩衝液(pH9.0,25mM)混合後,加入1.25g甘氨酸,室溫震蕩反應17h。而後加入1g NaBH4終止反應,將製得的微球用去離子水透析,並超濾濃縮至5%(w/w)的水溶液。製得羧基化PS/cPGMA微球,經電位滴定法測定羧基密度為0.150mM/g(幹球)。
分別取上述製得的羧基化PS/cPGMA微球與粒徑及羧基密度相同的羧基化PS微球各1mL,製備Cys-C LETIA檢測試劑。R1與R2的配方及製備方法如下:
R1:60mL,其中含有50mM PBS(pH7.4),0.5%BSA,0.05%NaN3,150mM NaCl。
R2:15mL,製備方法如下:取羧基化微球溶液或羧基化PS/cPGMA微球1mL,與1mL 50mM 2-嗎啉代乙磺酸(MES)緩衝液(pH6.0)混合後,加入EDC與N-羥基硫代琥珀醯亞胺各10mg後,室溫震蕩反應20min。加入10mg Cys-C多克隆抗體(產自瑞安市伊普西隆生物科技有限公司),37℃水浴震蕩反應3h。隨後,加入0.5g甘氨酸終止反應。將上述反應液用截留分子量為500kD的膜包切向流過濾除去未偶聯的抗體,所用的洗液為50mM PBS(pH7.4),切向流過濾後收集微球溶液15mL。最後向微球溶液中加入BSA 0.3g,製得R2。
Cys-C標準品穩定性的測定:對8mg/L,4mg/L,1mg/L標準品進行測試,採用加速試驗的方法進行穩定性測試。即:將上述製備的試劑均分為14份,完全密封后放入37℃水浴中,每天取出1份對標準品進行測試,連續測試14天。測試結果參見圖3。
由圖可知,採用PS/cPGMA納米粒製備的試劑,考察期間內,測試結果無明顯變化趨勢,標準品的信號值相對標準偏差(RSD)小於2.3%(圖3A);採用PS納米粒製備的試劑,考察期間內,測試結果有顯著下降趨勢,標準品的信號值相對標準偏差(RSD)顯著增大(圖3A)。這充分說明PS/cPGMA製備的LETIA檢測試劑比PS納米粒製備的檢測試劑具有更好的穩定性。
實施例5
將實施例1製備的PS/cPGMA納米粒和PS/PGMA納米粒(根據段殷等,單分散PS/PGMA核-殼型高分子微球的製備與表徵。天津醫科大學學報,2012,18:416-418.中的方法製備)的溶脹性進行比較。
將兩種納米粒均用50mM PBS(pH7.4,含0.05%NaN3)溶液配製成5%(w/w)的微球水溶液,並將其密封后放置在37℃水域中,測定21天 內的粒徑變化情況,對比結果參見圖4。
由圖4可知,PS/PGMA納米粒粒徑顯著增大(從第一天測定的120.4nm增大到第21天的167.8nm),而PS/cGMA納米粒的粒徑並未見明顯變化(第一天測得的粒徑為120.2nm,而到第21天的為120.0nm)。這說明本專利製備的納米粒穩定性得到了顯著提高。