一種鹽酸替匹嘧啶的晶型及其製備方法與流程
2023-12-10 05:57:32 4
本發明涉及晶型藥物領域,尤其涉及一種鹽酸替匹嘧啶的晶型及其製備方法。
背景技術:
鹽酸替匹嘧啶,商品名tipiracil(也稱tpi),結構如式i,tpi作為曲氟尿苷(也稱ftd)降解酶胸苷磷酸化酶的抑制劑,與ftd按摩爾比1:2聯合用藥,用於治療不可切除性或復發性晚期結直腸癌。其化學名為5-氯-6-[(2-亞氨基吡咯烷-1-基)甲基]嘧啶-2,4(1h,3h)-二酮。
目前已報導的鹽酸替匹嘧啶晶型有三種,其製備方法分別為:
晶型i:(1)將按照w096/30246記載方法得到的鹽酸替匹嘧啶95.1g加熱溶於100ml6n鹽酸和220ml水的混合物中,在60℃熱過濾後,加入1280ml乙醇,將混合物通過加熱將溫度維持在60℃兩小時,然後冰冷,過濾得到晶型i產物89.3g。
或(2)將按照w096/30246記載方法得到的鹽酸替匹嘧啶加熱溶解於水中,在60℃熱過濾後,加入乙醇和(1)中得到的籽晶(晶型i),加熱將溫度維持在60℃兩小時,過濾得到晶型i產物。
晶型ii:(1)將按照w096/30246記載方法得到的鹽酸替匹嘧啶61.5g溶於50ml6n鹽酸和500ml水的混合物中,進行活性炭處理,過濾後濃縮,在室溫下加入200ml乙醇,過濾得到晶型ii產物57.9g。
或(2)將按照w096/30246記載方法得到的鹽酸替匹嘧啶加入水中,在60℃下溶解。過濾後,加入到冰冷後的乙醇中,濾得到晶型ii產物。
晶型iii:(1)將按照w096/30246記載方法得到的鹽酸替匹嘧啶22.0g溶解於20ml6n鹽酸和230ml水中,過濾後濃縮,在室溫下加入100ml乙醇,過濾得到晶型iii產物19.7g
或(2)將按照w096/30246記載方法得到的鹽酸替匹嘧啶溶解於甲醇中,投入濃鹽酸。在64℃下攪拌1小時後,冷卻到30℃,過濾後用甲醇洗滌得到晶型iii產物。
對於藥物分子,不同的晶型結構可能具有不同的理化性質,包括表觀溶解度、溶解速率、穩定性、密度等。這些性質將對api的生產、製劑以及藥物的吸收等方面產生直接的影響。由於上述原因,對原料藥新晶型的發現和研究具有非常重大的實際意義。
技術實現要素:
有鑑於此,本發明要解決的技術問題在於提供一種鹽酸替匹嘧啶的晶型及其製備方法,本發明提供的鹽酸替匹嘧啶晶型溶解度高,其製備方法所得產品收率和純度皆高。
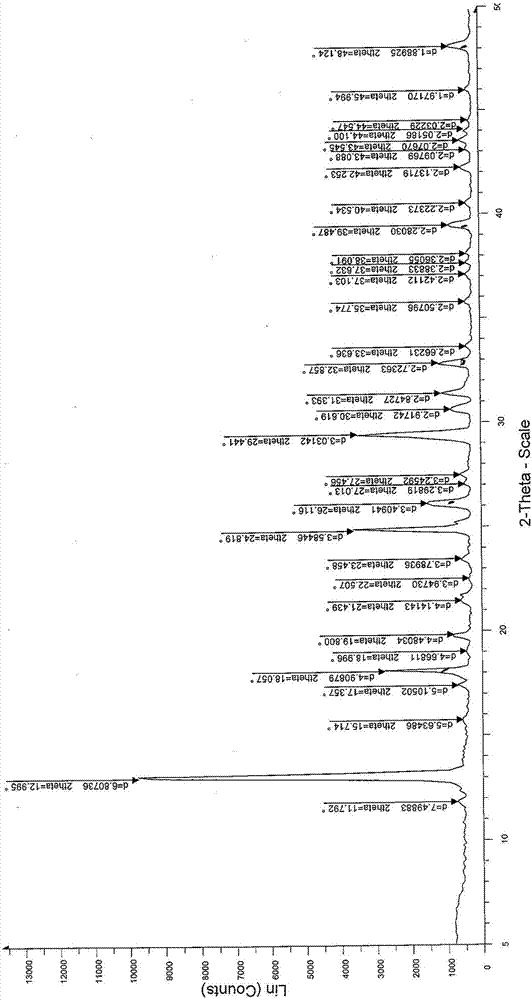
本發明提供的鹽酸替匹嘧啶的晶型,x-射線粉末衍射圖在2θ±0.2°位置有衍射峰,所述2θ為13.0°、18.1°、24.8°、26.1°、29.4°。
本發明提供的鹽酸替匹嘧啶晶型的2θ為13.0°、18.1°、19.8°、24.8°、26.1°、29.4°、30.6°、31.4°、32.9°、39.5°、48.1。
本發明提供的鹽酸替匹嘧啶晶型的2θ為13.0°、17.4°、18.1°、19.8°、21.4°、23.5°、24.8°、26.1°、27.5°、29.4°、30.6°、31.4°、32.9°、35.8°、39.5°、42.3°、43.5°、48.1。
根據x-射線粉末衍射圖,本發明所述的鹽酸替匹嘧啶晶型的x-射線衍射的詳細參數如表1:
表1本發明所述的鹽酸替匹嘧啶晶型x射線衍射參數
x-射線粉末衍射圖的2θ值可在機器之間或樣品之間稍有變化,其數值可能相差大約0.2個單位,或者相差大約0.1個單位,因此所引用的數值不能解釋為絕對值。同樣應該理解,峰高的大小同樣可能相差大約5個單位,或者相差大約4個單位,或者相差大約3個單位,或者相差大約2個單位,或者相差大約1個單位,因此包括於本發明中的xrpd跡線(trace)強度為說明性的,並非意欲用於絕對比較。
x-射線粉末衍射圖顯示,本發明提供的鹽酸替匹嘧啶晶型與現有報導的晶型i、晶型ii或晶型iii的2θ值皆不同,峰高值也不同,本發明提供的為一種新的晶型,命名為晶型j。本發明提供的鹽酸替匹嘧啶晶型與現有晶型相比,在化學穩定性、溶解性、生物活性方面皆存在一定的優勢,且本發明製備的鹽酸替匹嘧啶晶型j中雜質的含量較低而且收率較高。
本發明提供的鹽酸替匹嘧啶晶型的製備方法,包括:
將鹽酸替匹嘧啶與水混合,加熱至80℃~100℃溶解後,過濾;
60℃,向濾液滴加極性非質子溶劑至出現渾濁,降溫至15℃~25℃。攪拌1~2小時,過濾、乾燥獲得鹽酸替匹嘧啶晶型j。
影響鹽酸替匹嘧啶產生不同晶型的主要因素來自於各種物理條件參數的變化,例如,結晶時的溫度、晶體生長的時間,晶體生長的條件(攪拌或靜置)、重結晶的溶劑、溶劑與鹽酸替匹嘧啶的比例等。
在本發明中,鹽酸替匹嘧啶與水的質量體積比為60g:120ml。
一些實施例中,極性非質子溶劑為四氫呋喃、丙酮或n,n-二甲基甲醯胺。
一些實施例中,所述極性非質子溶劑與濾液的體積比為120:(60~360)。
一些具體實施例中,極性非質子溶劑為四氫呋喃,四氫呋喃與濾液的體積比為2:1。
該實施例中,鹽酸替匹嘧啶晶型j的製備方法為:鹽酸替匹嘧啶加熱溶解於水(質量-體積比為1g:2ml)中,80℃熱過濾後。維持60℃,向體系中滴加四氫呋喃(四氫呋喃與濾液體積比為2:1)後,體系緩慢降溫至15~25℃。攪拌1~2小時,過濾;所得固體經真空乾燥,得鹽酸替匹嘧啶晶型l。收率為:80.3%。
一些具體實施例中,極性非質子溶劑為丙酮,丙酮與濾液的體積比為3:1。
該實施例中,鹽酸替匹嘧啶晶型j的製備方法為:鹽酸替匹嘧啶加熱溶解於水(質量-體積比為1g:2ml)中,80℃熱過濾後。維持60℃,向體系中滴加丙酮(丙酮與濾液體積比為3:1)後,體系緩慢降溫至15~25℃。攪拌1~2小時,過濾;所得固體經真空乾燥,得鹽酸替匹嘧啶晶型l。收率為:66.5%。
一些具體實施例中,極性非質子溶劑為丙酮,丙酮與濾液的體積比為1:2。
該實施例中,鹽酸替匹嘧啶晶型j的製備方法為:鹽酸替匹嘧啶加熱溶解於水(質量-體積比為1g:2ml)中,80℃熱過濾後。維持60℃,向體系中滴加丙酮(丙酮與濾液體積比為1:2)後,體系緩慢降溫至15~25℃。攪拌1~2小時,過濾;所得固體經真空乾燥,得鹽酸替匹嘧啶晶型l。收率為:31.3%。
本發明中所述降溫為緩慢降溫,降溫的速率為2~3℃/min。
本發明提供的晶型j在製備治療不可切除性或復發性晚期結直腸癌的藥物中的應用。
本發明還提供了一種治療不可切除性或復發性晚期結直腸癌的藥物,其包括鹽酸替匹嘧啶晶型j及藥學上可接受的輔料。
本發明提供的鹽酸替匹嘧啶的晶型,x-射線粉末衍射圖在2θ±0.2°位置有衍射峰,所述2θ為13.0°、18.1°、24.8°、26.1°、29.4。鹽酸替匹嘧啶的這一晶型與現有報導中的晶型皆不一致,該晶型溶解度、穩定性和生物利用度與現有晶型相比皆存在一定的優勢。且該本發明提供的該晶型的製備方法簡單,適宜條件下收率和純度皆較高。
附圖說明
圖1示實施例1製得產品的x-粉末衍射圖譜。
具體實施方式
本發明提供了一種鹽酸替匹嘧啶的晶型及其製備方法,本領域技術人員可以借鑑本文內容,適當改進工藝參數實現。特別需要指出的是,所有類似的替換和改動對本領域技術人員來說是顯而易見的,它們都被視為包括在本發明。本發明的方法及應用已經通過較佳實施例進行了描述,相關人員明顯能在不脫離本發明內容、精神和範圍內對本文的方法和應用進行改動或適當變更與組合,來實現和應用本發明技術。
本發明採用的儀器皆為普通市售品,皆可於市場購得。其中,所述鹽酸替匹嘧啶的製備方法參照申請號為201710117569.x的中國專利申請文件。具體步驟為:將6-甲基尿嘧啶與氧化銅在溶劑中進行氧化反應,得到6-甲醯基尿嘧啶;還原6-甲醯基尿嘧啶,得到6-羥甲基尿嘧啶;將6-羥甲基尿嘧啶經氯代反應,得到6-氯甲基尿嘧啶。
下面結合實施例,進一步闡述本發明:
實施例1
將按照申請號為201710117569.x的中國專利申請報導方法,製備鹽酸替匹嘧啶。將所得鹽酸替匹嘧啶60.0g加熱溶解於120ml水中,80℃熱過濾後。維持60℃,向體系中滴加240ml四氫呋喃。停止滴加,體系緩慢降溫至15~25℃。攪拌1~2小時,過濾。所得固體經真空乾燥,得48.2g鹽酸替匹嘧啶晶型l。收率為80.3%;純度為99.952%。該實施例的收率和純度皆顯著高於實施例2和實施例3,該效果具有統計學意義。該實施例製得晶型的x-粉末衍射圖譜如圖1。
實施例2
將按照申請號為201710117569.x的中國專利申請報導方法,製備鹽酸替匹嘧啶。將所得鹽酸替匹嘧啶60.0g加熱溶解於120ml水中,80℃熱過濾後。維持60℃,向體系中滴加360ml丙酮。停止滴加,體系緩慢降溫至15~25℃。攪拌1~2小時,過濾。所得固體經真空乾燥,得39.9g固體,經鑑定為鹽酸替匹嘧啶晶型l。收率為66.5%;純度為99.57%。
實施例3
將按照申請號為201710117569.x的中國專利申請報導方法,製備鹽酸替匹嘧啶。將所得鹽酸替匹嘧啶60.0g加熱溶解於120ml水中,80℃熱過濾後。維持60℃,向體系中滴加60ml丙酮。停止滴加,體系緩慢降溫至15~25℃。攪拌1~2小時,過濾。所得固體經真空乾燥,得18.8g固體,經鑑定為鹽酸替匹嘧啶晶型l。收率為31.3%;純度為99.938%。
以上僅是本發明的優選實施方式,應當指出,對於本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護範圍。