熔融法製備左旋奧拉西坦晶型II的方法與流程
2023-10-27 11:56:02 5
本發明涉及左旋奧拉西坦,具體涉及一種製備左旋奧拉西坦晶型ii的方法。
背景技術:
奧拉西坦(oxiracetam)為新一代腦代謝改善藥,吡咯烷酮類(環gabob)衍生物,吡拉西坦類似物,可促進磷醯膽鹼和鄰醯乙醇胺合成,促進腦代謝,通過血腦屏障對特異性中樞神經通路有刺激作用,改善智力和記憶。對腦血管病、腦損傷、腦瘤(術後)、顱內感染、痴呆、腦變性疾病等具有良好療效。適用於輕中度血管性痴呆、老年性痴呆以及腦外傷等症引起的記憶與智能障礙。奧拉西坦由義大利史克比切姆公司於1974年首次合成,1987年上市,對記憶尤其是思維的集中比吡拉西坦更好,且毒性較小,研究表明其左旋體的療效更好,左旋奧拉西坦結構如下:
為有效將左旋奧拉西坦開發成藥品,需要一種具有易於製造並且可接受的化學和物理穩定性的固態形式,以促進其加工與流通儲存。對於增強化合物的純度和穩定性而言,結晶固體形態一般優於非晶型形態。目前,製備左旋奧拉西坦晶型的方法是先將左旋奧拉西坦溶於溶劑,再提取;比如cn102249975a公開了一種左旋奧拉西坦晶型i的製備方法,以丙酮和水為溶劑,緩慢析出晶體;又如cn102558013a公開了一種左旋奧拉西坦晶型ii及其製備方法,左旋奧拉西坦經過冰水頂洗過後結晶得到晶型ii;再如cn103553997a公開了使用正丙醇為溶劑,再提取製得晶型iii。上述方法各自存在不少缺點和不足,如操作繁瑣,容易形成混合晶型,有機溶劑用量大,不經濟環保,且生產成本較高,而且這些方法收率都偏低。
技術實現要素:
本發明的目的是提供一種左旋奧拉西坦晶型ii的製備方法,該方法製備工藝簡單,所需時間短,製得的產品純度高。
本發明所採取的技術方案為:
一種左旋奧拉西坦晶型ii的製備方法,包括如下步驟:左旋奧拉西坦在135~180℃加熱熔融,充n2保護,然後快速冷卻至-25~-15℃,靜置直至析晶。
上述快速冷卻的方式優選液氮冷卻或低溫冷卻槽。
本發明製備得到的左旋奧拉西坦晶型ii在衍射角度2θ為10.669、13.25、13.847、14.198、16.729、17.934、18.746、18.816、20.273、20.413、21.431、21.617、21.663、23.38、24.324、24.415、26.069、26.107、27.901、28.621、28.925、29.449、29.484、31.702、36.516、37.685、39.721度處有衍射峰,與cn102558013a披露的晶型一致。
在熔融製備左旋奧拉西坦晶型ii的過程中,發明人發現,熔融溫度控制不好可能會導致混合晶型,冷卻方式與溫度控制不好也可能出現混合晶型,甚至出現難以製得晶型的結果。同時,原料的純度對左旋奧拉西坦晶型ii的製備也有一定的影響。
優選的,先採用以下路線製得高純度左旋奧拉西坦,然後通過熔融的方式製得左旋奧拉西坦晶型ii。
一種左旋奧拉西坦晶型ii的製備方法,先製得左旋奧拉西坦,然後用熔融法培養晶型ii,其特徵在於,製備左旋奧拉西坦的路線為:
採用以下步驟:
1)、先用甲醇將s-4-氯-3-羥基丁酸乙酯溶解,然後在40~60℃下加入疊氮化鈉反應4~6小時得到中間體i,其中s-4-氯-3-羥基丁酸乙酯與疊氮化鈉摩爾比為1:1~2;
2)用dmso將中間體i溶解,以金屬鈀為催化劑與氫氣進行還原反應得到中間體ii,反應溫度為40~50℃,反應時間為6~8小時;
3)用dmso將中間體ii溶解,碳酸鉀為催化劑,與溴乙酸叔丁酯反應6~8小時,反應溫度為40~60℃;所述中間體ii與溴乙酸叔丁酯的摩爾比為:1:1~2,中間體ii與所述碳酸鉀的摩爾比為:1:2~3;
4)用叔丁醇將中間體ⅲ溶解,在80~100℃條件下進行關環反應得到中間體iv,反應時間為7~9小時;
5)將中間體iv與氨水在20~30℃下反應10~15小時,得到左旋奧拉西坦粗品,所述中間體iv:氨的摩爾比為中間體iv:氨=1:12~15,以氨氣甲醇溶液中的氨計,所述氨水的濃度為25-28%;
6)將左旋奧拉西坦粗品加熱溶解於水中,活性炭脫色,過濾除去活性炭,減壓濃縮除水,當剩餘水量為加入產物重量2~3倍時停止濃縮,0~5℃低溫冷卻結晶,獲得左旋奧拉西坦;
7)將左旋奧拉西坦在140~160℃加熱熔融,充n2保護,然後用低溫冷卻槽快速冷卻至-25~-19℃,靜置直至析晶。
本發明開闢了一條全新的培養左旋奧拉西坦單晶的路線,以特定的條件進行熔融析晶,在不需要有機溶劑的條件下成功的製得了左旋奧拉西坦晶型ii,工藝簡單,成本低且對環境友好,從而大大推動了左旋奧拉西坦晶型的科學研究及工業化生產。本發明工藝時間短,整個晶型培養不超過6小時,相對於cn102558013a而言,製備效率大大提高。
本發明以s-4-氯-3-羥基丁酸酯和疊氮化鈉為起始原料製備得到的高純度左旋奧拉西坦,中間體及產物均不需要柱層析,成本低,操作簡便,適合工業化大生產;製得的左旋奧拉西坦產物純度經高效液相檢測達到99.5%以上,而且製備過程中不產生難以去除的雜質然後用熔融法製備得到左旋奧拉西坦晶型ii,收率100%,光學純度在99.9%以上,大大的提高了左旋奧拉西坦晶型ii的產品質量;相對於cn102558013a而言,反應效率高,時間明顯縮短(本發明晶型培養不超過6小時,cn102558013a為1~3天。
附圖說明
圖1是左旋奧拉西坦水合物晶型ⅱ的粉末衍射圖;
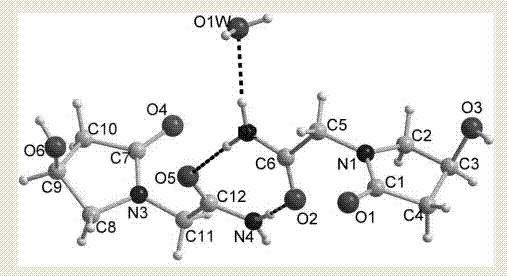
圖2是左旋奧拉西坦水合物晶型ⅱ的單晶結構圖;
圖3是左旋奧拉西坦水合物晶型ⅱ的晶胞堆積圖;
圖4是左旋奧拉西坦水合物晶型ⅱ的拉曼光譜圖;
圖5是左旋奧拉西坦水合物晶型ⅱ的熱重分析(tg)圖;
圖6是左旋奧拉西坦水合物晶型ⅱ的紅外光譜(ir)圖。
具體實施方式
下面通過實施例對本發明進行具體的描述,有必要在此指出的是以下實施例只用於對本發明進行進一步說明,不能理解為對本發明保護範圍的限制,該領域的技術人員可以根據上述本發明內容對本發明作出一些非本質的改進和調整。本發明所用原料與試劑均為市售產品。
實施例1
(l)中間體i的製備:
取原料s-4-氯-3-羥基丁酸乙酯5g,加入一單頸瓶中,加入甲醇10ml,加入疊氮化鈉5g攪拌,保持溫度50℃左右反應4小時,停止反應得黃色溶液。加入水20ml,用乙酸乙酯20ml萃取,濃縮除去乙酸乙酯,得到黃色油狀物中間體i。經核磁檢測,中間體i為:1h-nmr(300mhz,cdcl3):δ1.42-1.73(m,2h)2.76-2.67(absystem,m,2h,),3.31-3.23(absystem,m,2h),3.75(s,3h),4.40(m,1h),3.70(s,1h).
中間體i為:
(2)中間體ii的製備
將步驟(1)獲得的中間體i溶解於50ml的dmso中,加入10%鈀碳催化劑1.2g,通入氫氣在45℃左右攪拌6小時,點板見原料反應完全,停止反應,濃縮除去溶劑得到淡黃色油狀物中間體ii。經核磁檢測,中間體ii:1h-nmr(300mhz,d2o):1.30(m,3h),2.76-2.67(absystem,m,2h,),3.31-3.23(absystem,m,2h),4.12(m,2h),4.40(m,1h),4.70(bs,3h)。
中間體ii為
(3)中間體ⅲ的製備
將步驟(2)獲得的中間體ii溶解於50ml的dmso中,在40℃左右,加入碳酸鉀17.3g(3eq),有大量固體生成,攪拌五分鐘,開始滴加溴乙酸叔丁酯9ml(2eq),滴加過程有放熱現象,滴加完畢後繼續攪拌6小時,點板見原料反應完全,停止反應,加入ea(乙酸乙酯)50ml,水30ml,固體完全溶解,將水層用氯化鈉固體飽和,分出有機層,水層用ea20ml萃取兩次,合併有機層,有機層用2m的鹽酸20ml洗三次,合併鹽酸水相,有機相棄去,水相用碳酸氫鈉調節ph至8,固體氯化鈉飽和,ea30ml萃取三次,合併有機相,無水硫酸鎂乾燥,濃縮除去溶劑得到淡黃色油狀物,低溫下固化得到中間體ⅲ。經核磁檢測,中間體ⅲ:1h-nmr(300mhz,d2o):1.28-1.4(m,12h),2.28-2.53(m,2h),2.58-2.83(m,2h)3.51(s,2h),4.09-4.12(m,3h)。
中間體ⅲ為
(4)中間體iv的製備
將步驟(2)獲得的中間體ⅲ用50ml叔丁醇溶解,升溫至85℃反應8小時,得到一紅褐色溶液,點板見原料反應完全。停止反應,濃縮除去甲苯,加入ea(乙酸乙酯)溶解,過濾除去鹽,活性炭脫色,濃縮除去得黃色油狀物得到中間體iv。經核磁檢測,中間體iv為:1h-nmr(300mhz,cdcl3)1.402(m,9h),2.38(dd,1h),2.69(dd,1h),3.34(dd,1h),3.77(dd,lh),3.93(d,lh),4.18(d,1h),4.30(bs,1h),4.50(m,1h)。
中間體iv:
(5)左旋奧拉西坦的製備
將步驟(4)獲得的中間體iv加入濃氨水(濃度為26%)20ml,室溫攪拌14小時,點板見原料反應完全,停止反應,濃縮去除水和氨氣,得到黃色油狀物,加入丙酮溶解油狀物,加入少量晶種攪拌,析出固體,少量丙酮衝洗瓶壁,-10℃結晶5小時,過濾得到類白色粗品23.5g,化學純度99.0%。
(6)將該粗品溶解於100ml的水中,加熱使其溶解,活性炭脫色半小時,過濾除去活性炭,冷卻結晶,5℃放置過夜,次日過濾得白色固體21.8g,化學純度99.6%,終產物中不含有經核磁檢測,左旋奧拉西坦:1h-nmr(300mhz,dmso-d6)δ2.10(d,1h),2.57(dd,1h),3.69(d,1h),3.88(d,1h),4.10(d,1h),4.31(m,1h),5.25(s,1h),7.13(s,1h),7.33(s,1h)。
左旋奧拉西坦結構式如下:
(7)(s)-奧拉西坦晶型ii的製備:
將左旋奧拉西坦在155~160℃加熱熔融,充n2保護,然後用液氮快速冷卻至-19℃左右,靜置直至析晶。
將製得的左旋奧拉西坦晶型進行結構鑑定,圖1是左旋奧拉西坦水合物晶型ⅱ的粉末衍射圖;圖2是左旋奧拉西坦水合物晶型ⅱ的單晶結構圖;圖3是左旋奧拉西坦水合物晶型ⅱ的晶胞堆積圖;圖4是左旋奧拉西坦水合物晶型ⅱ的拉曼光譜圖;圖5是左旋奧拉西坦水合物晶型ⅱ的熱重分析(tg)圖;圖6是左旋奧拉西坦水合物晶型ⅱ的紅外光譜(ir)圖。
得到的左旋奧拉西坦晶型在衍射角度2θ為10.669、13.25、13.847、14.198、16.729、17.934、18.746、18.816、20.273、20.413、21.431、21.617、21.663、23.38、24.324、24.415、26.069、26.107、27.901、28.621、28.925、29.449、29.484、31.702、36.516、37.685、39.721度處有衍射峰,與cn102558013a披露的晶型ii一致。
得到的左旋奧拉西坦晶型ii產生的紅外光譜在以下波數顯示出吸收峰:
3318(cm-1)、3223(cm-1)、2929(cm-1)、2875(cm-1)、1680(cm-1)、1487(cm-1)、1402(cm-1)、1276(cm-1)、1220(cm-1)、1078(cm-1)、968(cm-1)、943(cm-1)、694(cm-1)、611(cm-1)。
將實施例1製得的左旋奧拉西坦晶型ii進行光學純度測定:
取左旋奧拉西坦晶型ii,精密稱取數量(相當於含左旋奧拉西坦120mg)置100ml量瓶,加流動相超聲溶解並定容製成每1ml中含左旋奧拉西坦1.2mg的溶液,作為供試品溶液。
精密量取該溶液20ul,注入液相色譜儀,記錄色譜圖,按面積歸一化法計算未知雜質的含量。
測定所用的色譜條件為:
儀器:島津lc-2010aht高效液相色譜儀;
工作站名稱:lc-solutio;
色譜柱:agop(4.6×100mm,5μm);
流動相:乙腈:磷酸緩衝液(ph6.0)=15:85;
檢測波長:210nm;
流速:1ml/min;
柱溫35℃;
計算公式如下:
公式中,ai為主藥活性成分左旋奧拉西坦的峰面積;
∑a為s-構型和r-異構體奧拉西坦的峰面積之和。
經三次測量,取平均值,得實施例1的左旋奧拉西坦晶型ii的光學純度為99.95%。
在熔融製備左旋奧拉西坦晶型ii的過程中,發明人發現,熔融溫度控制不好可能會導致混合晶型,冷卻方式與溫度控制不好也可能出現混合晶型,甚至出現難以製得晶型的結果。
對比例1
將5g左旋奧拉西坦在230℃左右加熱熔融,充n2保護,然後用低溫冷卻槽快速冷卻至-19℃左右,靜置直至析晶,經過x粉末衍射檢測,發現為晶型i與晶型ii的混合物。晶型i具體參數參見cn102249975a。
對比例2
將5g左旋奧拉西坦在150℃左右加熱熔融,空氣氛圍,然後用低溫冷卻槽快速冷卻至-19℃左右,靜置直至析晶,經過x粉末衍射檢測,發現為晶型ii,收率68%,經檢測有雜質包裹,難以成功分離。
對比例3
將5g左旋奧拉西坦在150℃左右加熱熔融,充n2保護,然後緩慢降溫,置於室溫析晶,經過x粉末衍射檢測,發現為晶型i與晶型ii的混合物。
參照實施例1製得實施例2-4,部分參數按照以下運行:
實施例2-4製得的(s)-奧拉西坦化學純度高,其中不含有常規純化(不經過柱層析處理)即可實現化學純度99.5%以上,用作原料參照實施例1的晶型培養,收率100%,其光學純度大於99.90%。