標記物質、免疫學的測定法、免疫學的測定用試藥、分析物的測定方法、分析物測定用套組以及側流型色譜用測試條與流程
2023-10-31 11:07:16 1
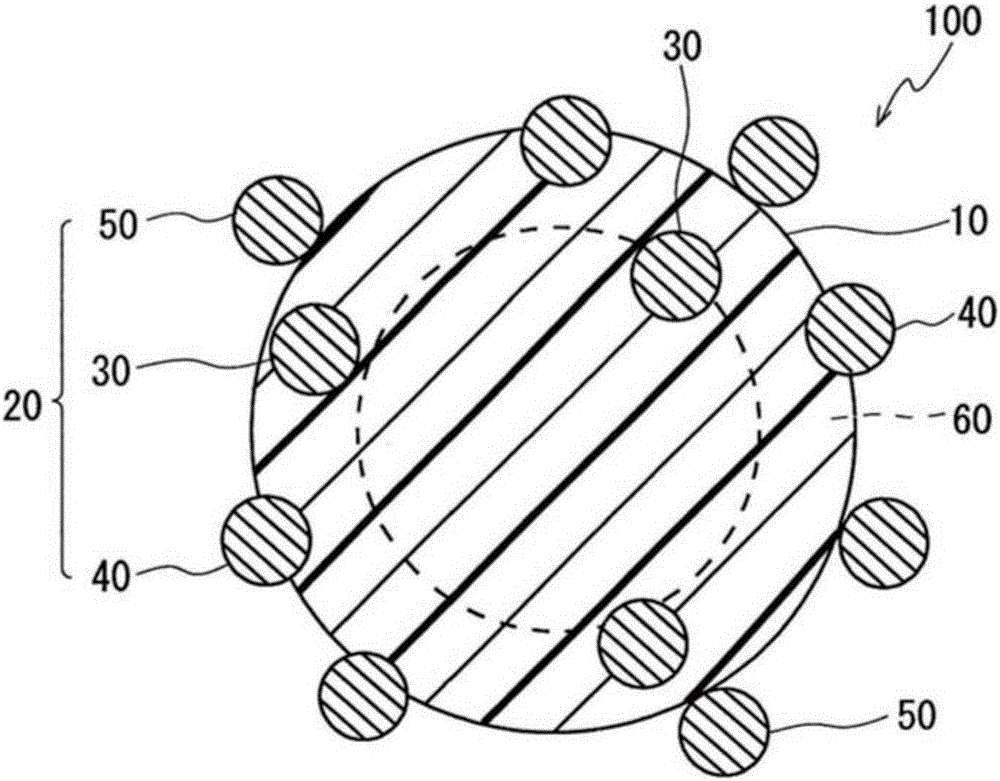
本發明涉及一種可用於免疫學的測定,感度、耐久性及視認性優異的標記物質及利用其的免疫學的測定法,免疫學的測定用試藥,分析物的測定方法,分析物測定用套組以及側流型色譜用測試條。
背景技術:
:在生物體內存在無數的化學物質,因此定性地、定量地對生物體內的特定的微量成分進行分析是極其重要的技術。在醫療、製藥、健康食品、生物技術、環境等領域中,僅對生物體內的特定的部位(化學物質)發揮作用的藥品及食品、檢測生物體的略微的變化的分析裝置及診斷藥等與所述技術一同發展。所述分析技術之一有免疫測定(immunoassay)。所述技術也被稱為免疫學的測定法,其是利用作為免疫反應之一的抗原-抗體間的特異的反應,定性地、定量地對微量成分進行分析的方法。抗原-抗體間反應因感度或反應的選擇性高,故廣泛地用於所述領域。免疫測定根據其測定原理而存在各種測定法。例如可列舉:酶免疫測定法(EnzymeImmunoassay,EIA)、放射性免疫測定法(Radioimmunoassay,RIA)、化學發光免疫測定法(ChemiluminescentImmunoassay,CLIA)、螢光免疫測定法(FluorescenceImmunoassay,FIA)、乳膠等的凝聚法(乳膠免疫測定法(LatexImmunoassay,LIA)、顆粒凝聚(ParticleAgglutination,PA))、免疫色譜法(Immunochromatography,ICA)、紅血球凝聚法(Hemagglutination,HA)、紅血球凝聚抑制法(HemagglutinationInhibition,HI)等。再者,除免疫測定以外,有物理·化學的測定法、生物學的測定法等。免疫測定根據抗原及抗體進行反應而形成複合體時的變化(抗原、抗體或複合體的濃度變化),定性地或定量地檢測抗原或抗體。當檢測這些時,使抗體、抗原或複合體與標記物質結合,由此檢測感度增大。因此,標記物質的標記能力可以說是影響免疫測定中的檢測能力的重要的要素。在以上所例示的免疫測定中,也使用紅血球(HA的情況)、乳膠粒子(LIA的情況)、螢光色素(FIA的情況)、放射性元素(RIA的情況)、酶(EIA的情況)、化學發光物質(CLIA的情況)等作為標記物質。然而,當使用經著色的微粒子作為標記物質時,可不使用特別的分析裝置而通過目視來確認檢測,因此期待可實現更簡便的測定。作為此種經著色的微粒子,可列舉:金屬及金屬氧化物的膠體狀粒子、利用色素進行了著色的乳膠粒子等(專利文獻1、專利文獻4等)。但是,所述膠體狀粒子根據粒徑及製備條件來決定色調,因此存在難以獲得所期望的鮮明的濃色調的,即視認性不充分這一問題。另外,所述經著色的乳膠粒子存在利用色素的著色的效果低、目視判定性不充分這一問題。再者,若為了消除所述問題而增加色素的著色量,則色素覆蓋乳膠的表面,乳膠粒子原本的表面狀態受損,因此存在難以使抗原或抗體結合這一問題。另外,還存在如下的問題:在薄膜過濾器等色譜介質的細孔內堵塞、或乳膠粒子產生非特異凝聚、或因增加色素的著色量而濃濃地著色的情況未必與性能的提升有關聯。為了提升所述標記物質的視認性,揭示有如下的免疫色譜方法:在結合有標記物質的抗體(標記抗體)與抗原進行反應而形成複合體後,進而使其他金屬對這些標記物質進行修飾,由此使標記物質的檢測感度增幅(專利文獻2及專利文獻5)。但是,在所述方法中,為了使金屬銀進行修飾而需要特別的裝置。因此,操作繁雜,難以實現穩定的增幅。另外,可認為因需要特別的裝置等而耗費測定成本,故可應用的用途及使用環境受到限定。另外,揭示有一種包含與聚合物系乳膠粒子的表面結合的金納米粒子的著色乳膠(專利文獻3)。通過使聚合物系乳膠粒子的表面與金納米粒子結合,所述金納米粒子本身作為著色劑而有助於目視判定性或檢測感度的提升,另一方面,金納米粒子本身對於抗原或抗體的結合性也優異,因此即便使金納米粒子結合至變成足夠濃的顏色的程度為止,也可使足夠量的抗原或抗體進行結合。所述著色乳膠是通過對苯乙烯-丙烯酸共聚物乳膠及金納米粒子的前體即HAuCl的分散液照射γ射線來使所述乳膠的表面與金納米粒子結合而成的。但是,所述著色乳膠因金納米粒子僅與乳膠的表面結合,故不僅顯現表面等離子體激元(surfaceplasmon)吸收的金粒子的承載量有限,而且金納米粒子容易脫離。其結果,存在作為免疫學的測定用試藥的視認性或感度不充分之虞。另外,因照射γ射線等電磁放射線,故存在對乳膠造成損害之虞。進而,在專利文獻3的說明書中揭示有所述乳膠徑或金納米粒子徑的優選的範圍,但在實施例中是否在這些優選的範圍內得到驗證並不明確,不存在優選範圍的規定依據。另外,在專利文獻4中揭示有一種由金屬金包覆的聚合物乳膠粒子,且暗示對於可用於顯微鏡檢査法及免疫測定法的試藥的應用。但是,所述由金屬金包覆的聚合物乳膠粒子未揭示聚合物乳膠粒子的材質或粒徑。進而,作為可用於免疫測定法的試藥的效果並無驗證。因此,作為金屬金及聚合物乳膠粒子中的試藥的效果不明。根據以上所述,結合或包覆有金納米粒子的乳膠粒子雖然作為免疫學的測定用的試藥而受到期待,但在現有的技術中,耐久性或視認性並不充分。另外,即便是視認性高的,可應用的用途及使用環境也受到限定。現有技術文獻專利文獻專利文獻1:日本專利特開平5-10950號公報專利文獻2:日本專利特開2011-117906號公報專利文獻3:日本專利特開2009-168495號公報專利文獻4:日本專利特開平3-206959號公報專利文獻5:日本專利特開2009-192270號公報技術實現要素:發明要解決的課題本發明的目的在於提供一種可用於免疫學的測定,感度、耐久性及視認性優異,且不需要特別的裝置或作業步驟的追加而可實現高感度的判定的標記物質。解決問題的技術手段本發明人等人進行努力研究的結果,發現通過利用具有特定的結構的樹脂-金屬複合體,可解決所述課題,從而完成了本發明。即,本發明的標記物質包括具有將金屬粒子固定於樹脂粒子上而成的結構的樹脂-金屬複合體,其特徵在於:具備以下的(A)、(B)的任一種構成:(A)所述樹脂-金屬複合體的平均粒徑超過300nm;或者(B)所述金屬粒子的平均粒徑為超過20nm、未滿70nm的範圍內。本發明的標記物質在所述(A)時,所述金屬粒子的平均粒徑可為1nm以上、80nm以下的範圍內。本發明的標記物質在所述(A)時,所述樹脂-金屬複合體的平均粒徑可為超過300nm、且為1000nm以下的範圍內。在此情況下,所述金屬粒子可為金粒子。本發明的標記物質在所述(B)時,所述樹脂-金屬複合體的平均粒徑可為100nm以上、1000nm以下的範圍內。在此情況下,所述金屬粒子可為金粒子。本發明的標記物質可為使所述樹脂-金屬複合體分散於水中而成者。本發明的標記物質可為使抗原或抗體吸附於所述樹脂-金屬複合體的表面來使用者。本發明的免疫學的測定法的特徵在於:使用所述任一種標記物質。本發明的免疫學的測定用試藥包括所述任一種標記物質。本發明的分析物的測定方法是對試樣中所含有的分析物進行檢測或定量的分析物的測定方法。本發明的分析物的測定方法使用包含薄膜、及將與所述分析物特異地結合的捕捉配體固定於所述薄膜上而成的判定部的側流型色譜用測試條。而且,本發明的分析物的測定方法的特徵在於:進行包括下述步驟(I)~步驟(III)的步驟;步驟(I):使試樣中所含有的所述分析物、與利用所述任一種標記物質對與所述分析物特異地結合的抗體進行標記而成的標記抗體接觸的步驟,步驟(II):在所述判定部中,使步驟(I)中所形成的含有分析物與標記抗體的複合體接觸捕捉配體的步驟,步驟(III):測定源自所述標記物質中的所述樹脂-金屬複合體的局部型表面等離子體激元共振的顯色強度的步驟。本發明的分析物測定用套組是使用側流型色譜用測試條,用以對試樣中所含有的分析物進行檢測或定量的分析物測定用套組。本發明的分析物測定用套組包括:側流型色譜用測試條,包含薄膜、及將與所述分析物特異地結合的捕捉配體固定於所述薄膜上而成的判定部;以及檢測試藥,包含利用根據權利要求第1項至第8項中任一項所述的標記物質對與所述分析物特異地結合的抗體進行標記而成的標記抗體。本發明的側流型色譜用測試條是用以對試樣中所含有的分析物進行檢測或定量的側流型色譜用測試條。本發明的側流型色譜用測試條包括:薄膜;判定部,在所述試樣展開的方向上,將與所述分析物特異地結合的捕捉配體固定於所述薄膜上而形成;以及反應部,在比所述判定部更上遊側包含利用根據權利要求第1項至第8項中任一項所述的標記物質對與所述分析物特異地結合的抗體進行標記而成的標記抗體。發明的效果本發明的標記物質具備具有將金屬粒子固定於樹脂粒子上而成的結構的樹脂-金屬複合體。因此,對於樹脂粒子的顯現局部型表面等離子體激元吸收的金屬粒子的承載量多。因此,本發明的標記物質作為耐久性及視認性優異、不需要特別的裝置或作業步驟的追加而可實現高感度的判定的優異的材料,可優選地應用於例如EIA、RIA、CLIA、FIA、LIA、PA、ICA、HA、HI等免疫學的測定。附圖說明圖1是表示構成本發明的一實施形態的標記物質的樹脂-金屬複合體的剖面的結構的示意圖。圖2是表示使用本發明的一實施形態的側流型色譜用測試條的分析物的測定方法的概要的說明圖。圖3是實施例1中所獲得的樹脂-金屬複合體的掃描型電子顯微鏡(ScanningElectronMicroscope,SEM)照片。圖4是實施例9中所獲得的樹脂-金複合體的掃描型電子顯微鏡(SEM)照片。圖5是實施例9中所獲得的樹脂-金複合體的剖面的掃描型透射電子顯微鏡(ScanningTransmissionElectronMicroscope,STEM)照片。圖6是實施例10中所獲得的樹脂-金複合體的掃描型電子顯微鏡(SEM)照片。圖7是實施例10中所獲得的樹脂-金複合體的剖面的掃描型透射電子顯微鏡(STEM)照片。具體實施方式以下,一面適宜參照附圖,一面對本發明的實施形態進行詳細說明。[第1實施形態]本發明的第1實施形態的標記物質具備具有將金屬粒子固定於樹脂粒子上而成的結構的樹脂-金屬複合體,且所述樹脂-金屬複合體的平均粒徑超過300nm。圖1是構成本實施形態的標記物質的樹脂-金屬複合體的剖面示意圖。樹脂-金屬複合體100具備樹脂粒子10與金屬粒子20。具有樹脂-金屬複合體100的本實施形態的標記物質例如可優選地用作免疫學的測定用試藥或其材料。樹脂-金屬複合體100將金屬粒子20分散或固定於樹脂粒子10上。另外,樹脂-金屬複合體100的金屬粒子20的一部分二維地或三維地分布於樹脂粒子10的表層部60中,且所述三維地分布的金屬粒子20的一部分朝樹脂粒子10外部分地露出,剩餘的一部分內包於樹脂粒子10中。此處,在金屬粒子20中存在完全地內包於樹脂粒子10中的金屬粒子(以下,也稱為「內包金屬粒子30」)、具有包埋於樹脂粒子10內的部位及朝樹脂粒子10外露出的部位的金屬粒子(以下,也稱為「部分露出金屬粒子40」))、及吸附於樹脂粒子10的表面的金屬粒子(以下,也稱為「表面吸附金屬粒子50」)。當將樹脂-金屬複合體100用作標記物質時,將抗體或抗原固定於部分露出金屬粒子40或表面吸附金屬粒子50上來使用。此時,將所述抗體或抗原固定於部分露出金屬粒子40及表面吸附金屬粒子50上,另一方面,不固定於內包金屬粒子30上。但是,包含內包金屬粒子30的金屬粒子20均顯現局部型表面等離子體激元吸收,因此不僅部分露出金屬粒子40及表面吸附金屬粒子50有助於標記物質的視認性提升,內包金屬粒子30也有助於標記物質的視認性提升。進而,與表面吸附金屬粒子50相比,部分露出金屬粒子40及內包金屬粒子30與樹脂粒子10的接觸面積大,除此以外,由埋包狀態所產生的定錨效應等物理的吸附力強,難以自樹脂粒子10中脫離。因此,可使作為使用樹脂-金屬複合體100的標記物質的耐久性、穩定性變得優異。內包金屬粒子30是整個表面被構成樹脂粒子10的樹脂覆蓋的。另外,部分露出金屬粒子40是其表面積的5%以上、未滿100%被構成樹脂粒子10的樹脂覆蓋的。就耐久性的觀點而言,其下限優選為表面積的20%以上,更優選為30%以上。另外,表面吸附金屬粒子50是其表面積的超過0%、未滿5%被構成樹脂粒子10的樹脂覆蓋的。另外,相對於樹脂-金屬複合體100的重量,對於樹脂-金屬複合體100的金屬粒子20(內包金屬粒子30、部分露出金屬粒子40及表面吸附金屬粒子50的合計)的承載量優選為5wt%(重量百分比)~70wt%。若為所述範圍,則樹脂-金屬複合體100的作為標記物質的視認性、目視判定性及檢測感度優異。若金屬粒子20的承載量未滿5wt%,則存在抗體或抗原的固定化量變少、檢測感度下降的傾向。金屬粒子20的承載量更優選為15wt%~70wt%。另外,優選為金屬粒子20的10wt%~90wt%為部分露出金屬粒子40及表面吸附金屬粒子50。若為所述範圍,則可充分確保朝金屬粒子20上的抗體或抗原的固定化量,因此作為標記物質的感度高。更優選為金屬粒子20的20wt%~80wt%為部分露出金屬粒子40及表面吸附金屬粒子50,就耐久性的觀點而言,進而更優選為表面吸附金屬粒子50為20wt%以下。另外,為了在免疫學的測定中獲得優異的檢測感度,優選為金屬粒子20的60wt%~100wt%、優選為75wt%~100wt%、更優選為85wt%~100wt%存在於表層部60中,更優選為存在於自樹脂粒子10的表面起在深度方向上為粒子半徑的40%的範圍內。另外,存在於表層部60中的金屬粒子20的5wt%~90wt%為部分露出金屬粒子40或表面吸附金屬粒子50,可充分確保朝金屬粒子20上的抗體或抗原的固定化量,因此作為標記物質的感度高,故優選。換言之,存在於表層部60中的金屬粒子20的10wt%~95wt%為內包金屬粒子30即可。此處,所述「表層部」是指以樹脂-金屬複合體100的最外側的位置(即,部分露出金屬粒子40或表面吸附金屬粒子50的突出端部)為基準,自樹脂粒子10的表面起在深度方向上為粒子半徑的50%的範圍。另外,所述「二維地分布」是指金屬粒子20分布於樹脂粒子10的面方向上。所述「三維地分布」是指金屬粒子20不僅分布於樹脂粒子10的面方向上,也分布於深度方向上。就金屬粒子20難以自樹脂粒子10中脫離的觀點及金屬粒子20的承載量變多的觀點而言,優選為金屬粒子20「三維地分布」。另外,在本實施形態中,樹脂-金屬複合體100的平均粒徑為超過300nm。若樹脂-金屬複合體100的平均粒徑為300nm以下,則存在作為標記物質的視認性或感度下降的傾向。樹脂-金屬複合體100的平均粒徑優選為超過300nm、且為1000nm以下,更優選為340nm以上、未滿650nm。此處,樹脂-金屬複合體100的粒徑是指樹脂粒子10的粒徑與部分露出金屬粒子40或表面吸附金屬粒子50的突出部位的長度相加所得的值,可利用雷射衍射/散射法、動態光散射法、或離心沉澱法進行測定。樹脂粒子10優選為結構中具有可吸附金屬離子的取代基的聚合物粒子。尤其,優選為含氮聚合物粒子。含氮聚合物中的氮原子容易化學吸附視認性優異、抗原或抗體的固定化容易的金、鈀等金屬粒子的前體即陰離子性金屬離子,故優選。在本實施形態中,使吸附於含氮聚合物中的金屬離子還原,而形成金屬納米粒子,因此所生成的金屬粒子20的一部分成為內包金屬粒子30或部分露出金屬粒子40。另外,如丙烯酸聚合體般,羧酸等可吸附陽離子性金屬離子,因此容易吸附銀、鎳、銅等金屬粒子的前體即陽離子性金屬離子,而可形成銀、鎳、銅等的金屬粒子20,也可製作與所述金、鈀等金屬的合金。另一方面,在為結構中具有可吸附金屬離子的取代基的含氮聚合物以外的樹脂粒子,例如聚苯乙烯等的情況下,樹脂內部難以吸附所述金屬離子。其結果,所生成的金屬粒子20的大部分成為表面吸附金屬粒子50。如上所述,表面吸附金屬粒子50與樹脂粒子10的接觸面積小,因此存在樹脂與金屬的接著力小、金屬粒子20自樹脂粒子10中脫離的影響大的傾向。所述含氮聚合物為主鏈或側鏈上具有氮原子的樹脂,例如有多胺、聚醯胺、多肽、聚氨基甲酸酯、聚脲、聚醯亞胺、聚咪唑、聚噁唑、聚吡咯、聚苯胺等。這些之中,優選為聚-2-乙烯基吡啶、聚-3-乙烯基吡啶、聚-4-乙烯基吡啶等多胺。另外,當在側鏈上具有氮原子時,例如可廣泛利用丙烯酸樹脂、酚樹脂、環氧樹脂等。金屬粒子20例如可應用銀、鎳、銅、金、鈀等。優選為視認性優異、抗原或抗體的固定化容易的金及鈀。這些顯現源自局部型表面等離子體激元共振的吸收,故優選。更優選為保存穩定性良好的金。這些金屬能夠以單體或合金等複合體來使用。此處,例如作為金合金,是指包含金與金以外的金屬種類,並含有10wt%以上的金的合金。另外,通過掃描型電子顯微鏡(SEM)觀察來測定長度的金屬粒子20的平均粒徑例如優選為1nm~80nm。在金屬粒子20的平均粒徑未滿1nm的情況或超過80nm的情況下,局部型表面等離子體激元難以顯現,因此存在感度下降的傾向。當金屬粒子20為金粒子時,第1實施形態中的金屬粒子20的平均粒徑優選為20nm以上、未滿70nm,更優選為22nm以上、未滿50nm。[第2實施形態]本發明的第2實施形態的標記物質具備具有將金屬粒子固定於樹脂粒子上而成的結構的樹脂-金屬複合體,且所述金屬粒子的平均粒徑為超過20nm、未滿70nm的範圍內。構成本實施形態的標記物質的樹脂-金屬複合體除金屬粒子的平均粒徑的範圍、及樹脂-金屬複合體的平均粒徑的範圍與第1實施形態(圖1)的樹脂-金屬複合體100不同以外,與第1實施形態(圖1)的樹脂-金屬複合體100相同。以下,也一面參照圖1,一面以與第1實施形態的不同點為中心進行說明。在第2實施形態中所使用的樹脂-金屬複合體100中,通過掃描型電子顯微鏡(SEM)觀察來測定長度的金屬粒子20的平均粒徑例如超過20nm、未滿70nm。若金屬粒子20的平均粒徑為20nm以下,則存在感度下降的傾向,若為70nm以上,則存在視認性下降的傾向。金屬粒子20的平均粒徑更優選為22nm以上、未滿50nm。另外,樹脂-金屬複合體100的平均粒徑例如優選為100nm~1000nm。若樹脂-金屬複合體100的平均粒徑未滿100nm,則例如當將金粒子用作金屬粒子20時,存在金粒子的承載量變少的傾向,因此存在與同尺寸的金粒子相比著色變弱的傾向,若超過1000nm,則存在當製成試藥時,容易在薄膜過濾器等色譜介質的細孔內堵塞的傾向、或分散性下降的傾向。樹脂-金屬複合體100的平均粒徑優選為100nm以上、未滿700nm,更優選為340nm以上、未滿650nm。此處,樹脂-金屬複合體100的粒徑是指樹脂粒子10的粒徑與部分露出金屬粒子40或表面吸附金屬粒子50的突出部位的長度相加所得的值,可利用雷射衍射/散射法、動態光散射法、或離心沉澱法進行測定。在第2實施形態的標記物質中所使用的樹脂-金屬複合體100中,所述以外的構成與第1實施形態中所使用的樹脂-金屬複合體100相同,故省略說明。[樹脂-金屬複合體的製造方法]所述第1實施形態及第2實施形態的標記物質中所使用的樹脂-金屬複合體100的製造方法並無特別限定。例如,向利用乳化聚合法所製造的樹脂粒子10的分散液中添加含有金屬離子的溶液,使金屬離子吸附於樹脂粒子10上(以下,稱為「金屬離子吸附樹脂粒子」)。進而,將金屬離子吸附樹脂粒子添加至還原劑溶液中,由此使金屬離子還原而生成金屬粒子20,從而獲得樹脂-金屬複合體100。另外,例如當使用金粒子作為金屬粒子20時,作為含有金屬離子的溶液,可列舉氯金酸(chloroauricacid)(HAuCl4)水溶液等。另外,還可使用金屬絡合物來代替金屬離子。另外,作為含有金屬離子的溶液的溶媒,可使用甲醇、乙醇、正丙醇、異丙醇、正丁醇、仲丁醇、叔丁醇等含水醇或醇,鹽酸、硫酸、硝酸等酸等來代替水。另外,視需要,也可向所述溶液中添加例如聚乙烯醇等水溶性高分子化合物、表面活性劑、醇類;四氫呋喃、二乙醚、二異丙醚等醚類;烷二醇、聚烷二醇、這些的單烷基醚或二烷基醚、甘油等多元醇類;丙酮、甲基乙基酮等酮類等各種水混合性有機溶媒等添加劑。此種添加劑促進金屬離子的還原反應速度,另外,對於控制所生成的金屬粒子20的大小有效。另外,還原劑可使用公知的。例如可列舉:硼氫化鈉、二甲基胺硼烷、檸檬酸、次亞磷酸鈉、水合肼(hydrazinehydrate)、鹽酸肼(hydrazinehydrochloride)、硫酸肼(hydrazinesulfate)、甲醛、蔗糖、葡萄糖、抗壞血酸、次膦酸鈉、對苯二酚、羅謝耳鹽(rochellesalt)等。其中,優選為硼氫化鈉或二甲基胺硼烷、檸檬酸。在還原劑溶液中,視需要可添加表面活性劑、或調整溶液的pH。pH調整可利用硼酸或磷酸等緩衝劑,鹽酸或硫酸等酸,氫氧化鈉或氫氧化鉀等鹼來調整。進而,利用還原劑溶液的溫度來調整金屬離子的還原速度,由此可控制所形成的金屬粒子的粒徑。另外,當使所述金屬離子吸附樹脂粒子中的金屬離子還原來生成金屬粒子20時,可將所述金屬離子吸附樹脂粒子添加至還原劑溶液中,也可將還原劑添加至所述金屬離子吸附樹脂粒子中,但就內包金屬粒子30及部分露出金屬粒子40的生成容易性的觀點而言,優選為前者。另外,為了保持樹脂-金屬複合體100對於水的分散性,例如可添加檸檬酸、聚-L-賴氨酸、聚乙烯基吡咯烷酮(polyvinylpyrrolidone)、聚乙烯基吡啶、聚乙烯醇、迪斯帕畢克(DISPERBYK)194、迪斯帕畢克(DISPERBYK)180、迪斯帕畢克(DISPERBYK)184(日本畢克化學(BYK-ChemieJapan)公司製造)等分散劑。進而,可利用硼酸或磷酸等緩衝劑,鹽酸或硫酸等酸,氫氧化鈉或氫氧化鉀等鹼來調整pH,並保持分散性。具有以上的構成的樹脂-金屬複合體100尤其通過使抗原或抗體吸附於金屬粒子20的表面而可優選地作為標記物質應用於例如EIA、RIA、CLIA、FIA、LIA、PA、ICA、HA、HI等免疫學的測定法。另外,尤其可優選地用作低濃度區域(高感度區域)中的目視判定性優異的標記物質。另外,標記物質的形態並無特別限定,例如,可用作使樹脂-金屬複合體100分散於水或調整了pH的緩衝液中而成的分散液。作為使抗原或抗體吸附於所述金屬粒子20的表面的方法,並無特別限定,可使用利用公知的物理吸附及化學吸附的方法。例如可列舉:使樹脂-金屬複合體100浸漬於含有抗原或抗體的緩衝液中進行培養(incubate)等物理吸附,或將SH基導入至抗原或抗體中,並與樹脂-金屬複合體100進行反應而形成Au-SH鍵等化學吸附。其中,就金屬粒子20與抗原或抗體的結合變得牢固而言,優選為化學吸附。其次,對將樹脂-金屬複合體100用作標記物質的分析物的測定方法、側流型色譜用測試條及分析物檢測·定量套組進行說明。[側流型色譜用測試條]首先,一面參照圖2,一面對本發明的一實施形態的側流型色譜用測試條(測試條)進行說明。如後述般,所述測試條200可優選地用於本發明的一實施形態的分析物的測定方法。測試條200具備薄膜110。在薄膜110中,在試樣的展開方向上依次設置有試樣添加部120、判定部130及吸液部140。<薄膜>作為測試條200中所使用的薄膜110,可應用一般的測試條中用作薄膜材料的。薄膜110例如由包含如下的微細多孔性物質的惰性物質(不與分析物160、各種配體等進行反應的物質)形成,所述微細多孔性物質顯示出毛細管現象,添加試樣的同時,試樣展開。作為薄膜110的具體例,可列舉:包含聚氨基甲酸酯、聚酯、聚乙烯、聚氯乙烯、聚偏二氟乙烯(polyvinylidenefluoride)、尼龍、纖維素衍生物等的纖維狀或不織纖維狀基質、膜、濾紙、玻璃纖維濾紙、布、棉等。這些之中,優選為可使用包含纖維素衍生物或尼龍的膜、濾紙、玻璃纖維濾紙等,更優選為可使用硝基纖維素膜、混合硝基纖維素酯(硝基纖維素與乙酸纖維素的混合物)膜、尼龍膜、濾紙。為了使操作變得更簡便,測試條200優選為具備支撐薄膜110的支撐體。作為支撐體,例如可使用塑料等。<試樣添加部>測試條200也可具有用以添加含有分析物160的試樣的試樣添加部120。在測試條200中,試樣添加部120是用以接收含有分析物160的試樣的部位。試樣添加部120可在試樣展開的方向上,形成在比判定部130更上遊側的薄膜110上,或者也可在薄膜110上設置例如包含纖維素濾紙、玻璃纖維、聚氨基甲酸酯、聚乙酸酯、乙酸纖維素、尼龍、棉布等材料的試樣添加墊來構成試樣添加部120。<判定部>在判定部130中固定有與分析物160特異地結合的捕捉配體131。捕捉配體131隻要是與分析物160形成特異的結合的,則可無特別限制地使用,例如可優選地使用針對分析物160的抗體等。即便在向測試條200提供試樣的情況下,捕捉配體131也以不自判定部130移動的方式固定化。捕捉配體131隻要通過物理的結合或化學的結合或者吸附等,而直接地或間接地固定於薄膜110上即可。另外,判定部130隻要是如含有標記抗體150與分析物160的複合體170接觸與分析物160特異地結合的捕捉配體131般的構成,則並無特別限定。例如,可在薄膜110上直接固定捕捉配體131,或者也可在固定在薄膜110上的包含纖維素濾紙、玻璃纖維、不織布等的墊上固定捕捉配體131。<吸液部>吸液部140例如由纖維素濾紙、不織布、布、乙酸纖維素等吸水性材料的墊形成。所添加的試樣的展開前線(frontline)到達吸液部140後的試樣的移動速度根據吸液部140的材質、大小等而不同。因此,可通過吸液部140的材質、大小等的選定,而對分析物160的檢測·定量設定最合適的速度。再者,吸液部140為任意的構成,也可省略。測試條200視需要可進而含有反應部、控制部等任意的部位。<反應部>雖然省略圖示,但在測試條200中,也可在薄膜110上形成含有標記抗體150的反應部。反應部可在試樣流動的方向上,設置在比判定部130更上遊側。再者,也可將圖2中的試樣添加部120用作反應部。當測試條200具有反應部時,若向反應部或試樣添加部120提供含有分析物160的試樣,則在反應部中,可使試樣中所含有的分析物160與標記抗體150接觸。在此情況下,僅向反應部或試樣添加部120提供試樣,由此可形成含有分析物160與標記抗體150的複合體170,因此可實現所謂的一步驟型的免疫色譜法。反應部只要含有與分析物160特異地結合的標記抗體150,則並無特別限定,可為將標記抗體150直接塗布於薄膜110上而成的。或者,反應部也可為將使標記抗體150含浸於例如包含纖維素濾紙、玻璃纖維、不織布等的墊(結合墊(conjugatepad))中的固定在薄膜110上而成的。<控制部>雖然省略圖示,但測試條200也可在試樣展開的方向上,形成將與標記抗體150特異地結合的捕捉配體固定在薄膜110上而成的控制部。通過在判定部130與控制部中一同測定顯色強度,可確認向測試條200提供的試樣展開後到達反應部及判定部130,且檢査正常地進行。再者,控制部除使用與標記抗體150特異地結合的其他種類的捕捉配體來代替捕捉配體131以外,以與所述判定部130相同的方式製作,可採用相同的構成。[分析物的測定方法]其次,對使用測試條200所進行的本發明的一實施形態的分析物160的測定方法進行說明。本實施形態的分析物160的測定方法是對試樣中所含有的分析物160進行檢測或定量的分析物160的測定方法。實施形態的分析物160的測定方法可使用包含薄膜110、及將與分析物160特異地結合的捕捉配體131固定於所述薄膜110上而成的判定部130的測試條200,並包括下述步驟(I)~步驟(III);步驟(I):使試樣中所含有的所述分析物160、與利用具有將多個金屬粒子20固定於樹脂粒子10上而成的結構的樹脂-金屬複合體100對與所述分析物160特異地結合的抗體進行標記而成的標記抗體150接觸的步驟,步驟(II):在判定部130中,使步驟(I)中所形成的含有分析物160與標記抗體150的複合體接觸捕捉配體131的步驟,步驟(III):測定源自樹脂-金屬複合體100的局部型表面等離子體激元共振的顯色強度的步驟。步驟(I):步驟(I)是使試樣中所含有的分析物160接觸標記抗體150的步驟。只要形成含有分析物160與標記抗體150的複合體170,則接觸的形態並無特別限定。例如,可向測試條200的試樣添加部120或反應部(省略圖示)提供試樣,並在所述反應部中使分析物160接觸標記抗體150,也可在向測試條200提供試樣前,使試樣中的分析物160接觸標記抗體150。步驟(I)中所形成的複合體170在測試條200上展開後移動,併到達判定部130。步驟(II):步驟(II)是在測試條200的判定部130中,使步驟(I)中所形成的含有分析物160與標記抗體150的複合體170接觸捕捉配體131。若使複合體170接觸捕捉配體131,則捕捉配體131與複合體170的分析物160特異地結合。其結果,複合體170在判定部130中被捕捉。再者,捕捉配體131不與標記抗體150特異地結合,因此當未與分析物160結合的標記抗體150到達判定部130時,所述未與分析物160結合的標記抗體150通過判定部130。此處,當在測試條200中形成有固定有與標記抗體150特異地結合的其他捕捉配體的控制部(省略圖示)時,通過了判定部130的標記抗體150繼續展開,並在控制部中與所述其他捕捉配體結合。其結果,未與分析物160形成複合體170的標記抗體150在控制部中被捕捉。步驟(II)之後,視需要在步驟(III)之前,例如可實施利用水、生理食鹽水、磷酸緩衝液等生化學檢査中所通用的緩衝液,對測試條200進行清洗的清洗步驟。利用清洗步驟,可將未在判定部130、或判定部130及控制部中被捕捉的標記抗體150(未與分析物160結合,而未形成複合體170的標記抗體150)去除。通過實施清洗步驟,當在步驟(III)中,測定由判定部130、或判定部130及控制部中的樹脂-金屬複合體100的局部型表面等離子體激元共振所引起的顯色時,可降低背景的顯色強度,可提高信號/背景比,並可進一步提升檢測感度或定量性。步驟(III):步驟(III)是測定源自樹脂-金屬複合體100的局部型表面等離子體激元共振的顯色強度的步驟。在實施所述步驟(II)或視需要的清洗步驟後,在測試條200中,測定源自樹脂-金屬複合體100的局部型表面等離子體激元共振的顯色強度。再者,當在測試條200中形成有控制部時,利用步驟(II),在控制部中,標記抗體150由其他捕捉配體捕捉而形成複合體。因此,在步驟(III)中,在測試條200中,不僅可在判定部130中產生由局部型表面等離子體激元共振所引起的顯色,在控制部中也可產生由局部型表面等離子體激元共振所引起的顯色。如此,通過在判定部130與控制部中一同測定顯色強度,可確認向測試條200提供的試樣是否正常地展開後到達反應部及判定部130。<試樣及分析物>本實施形態的分析物的測定方法中的試樣只要是包含蛋白質等可成為抗原的物質作為分析物160的,則並無特別限定。例如可列舉:含有目標分析物160的生物體試樣(即,全血、血清、血漿、尿、唾液、咳痰、鼻腔拭子液或咽拭子液、脊髓液、羊水、乳頭分泌液、眼淚、汗、來自皮膚的浸出液、來自組織或細胞及糞便的提取液等)或食品的提取液等。視需要,為了容易產生標記抗體150及捕捉配體131與分析物160的特異的結合反應,也可在所述步驟(I)前,對試樣中所含有的分析物160進行前處理。此處,作為前處理,可列舉利用酸、鹼、表面活性劑等各種化學藥品等的化學的處理,或利用加熱·攪拌·超聲波等的物理的處理。尤其,當分析物160為流感病毒核蛋白(nucleoprotein,NP)抗原等通常未露出至表面的物質時,優選為利用表面活性劑等進行處理。作為用於所述目的的表面活性劑,可考慮特異的結合反應,例如抗原抗體反應等的配體與分析物160的結合反應性,而使用非離子性表面活性劑。另外,所述試樣可利用通常的免疫學的分析法中所使用的溶媒(水、生理食鹽水、或緩衝液等)或水混合有機溶媒來適宜稀釋。作為所述分析物160,例如可列舉腫瘤指標物、信號傳送物質、激素等的蛋白質(包含多肽、寡肽等)、核酸(包含單鏈或雙鏈的脫氧核糖核酸(Deoxyribonucleicacid,DNA)、核糖核酸(Ribonucleicacid,RNA)、多核苷酸(polynucleotide)、寡核苷酸、肽核酸(Peptidenucleicacid,PNA)等)或具有核酸的物質、糖(包含寡糖、多糖類、糖鏈等)或具有糖鏈的物質、脂質等其他分子,只要是與標記抗體150及捕捉配體131特異地結合的,則並無特別限定,例如可列舉:癌胚抗原(Carcino-EmbryonicAntigen,CEA)、HER2蛋白、前列腺特異抗原(ProstateSpecificAntigen,PSA)、CA19-9、α-胎蛋白(α-fetoprotein,AFP)、免疫抑制酸性蛋白(IPA)、CA15-3、CA125、雌激素受體、孕酮受體、糞隱血、肌鈣蛋白I、肌鈣蛋白T、CK-MB、CRP、人絨毛膜促性腺激素(HumanChorionicGonadotrophin,HCG)、黃體形成激素(LuteinizingHormone,LH)、卵泡刺激激素(FollicleStimulatingHormone,FSH)、梅毒抗體、流感病毒人體血紅蛋白、衣原體抗原、A群β溶血性鏈球菌抗原、HBs抗體、HBs抗原、輪狀病毒、腺病毒、白蛋白、糖化白蛋白等。這些之中,優選為利用非離子性表面活性劑而可溶化的抗原,更優選為如病毒的核蛋白質般形成自集合體的抗原。<標記抗體>標記抗體150用於在步驟(I)中,接觸試樣中所含有的分析物160,而形成含有分析物160與標記抗體150的複合體170。標記抗體150為利用具有將多個金屬粒子20固定於樹脂粒子10上而成的結構的樹脂-金屬複合體100對與分析物160特異地結合的抗體進行標記化而成的。此處,所謂「標記化」,是指在步驟(I)~步驟(III)中,以樹脂-金屬複合體100不自標記抗體150中脫離的程度,通過化學的結合或物理的結合或者吸附等將樹脂-金屬複合體100直接地或間接地固定於抗體上。例如,標記抗體150可為抗體與樹脂-金屬複合體100直接結合而成的,也可為抗體與樹脂-金屬複合體100經由任意的連接分子結合而成的、或分別固定於不溶性粒子上而成的。另外,在本實施形態中,作為「抗體」,並無特別限制,例如除多株抗體、單株抗體、通過基因重組所獲得的抗體以外,可使用與抗原具有結合能力的抗體片段[例如H鏈、L鏈、Fab、F(ab')2等]等。另外,作為免疫球蛋白,可為IgG、IgM、IgA、IgE、IgD的任一種。作為抗體的產生動物種類,可以人為首、以及人以外的動物(例如小鼠、大鼠、兔子、山羊、馬等)。作為抗體的具體例,可列舉:抗PSA抗體、抗AFP抗體、抗CEA抗體、抗腺病毒抗體、抗流感病毒抗體、抗HCV抗體、抗IgG抗體、抗人IgE抗體等。<標記抗體的優選的製作方法>其次,列舉標記抗體150的優選的製作方法來進行說明。標記抗體150的製造至少可包含以下的步驟A;步驟A)在第1pH條件下將樹脂-金屬複合體100與抗體混合來進行結合,由此獲得標記抗體150的步驟,優選為進而包含步驟B;步驟B)在第2pH條件下對標記抗體150進行處理的步驟。[步驟A]在步驟A中,在第1pH條件下將樹脂-金屬複合體100與抗體混合而獲得標記抗體150。步驟A優選為使固體狀的樹脂-金屬複合體100以分散於液相中的狀態與抗體接觸。第1pH條件根據樹脂-金屬複合體100中的金屬粒子20的金屬種類而不同。當樹脂-金屬複合體100的金屬粒子20為金粒子(包含金合金粒子;以下相同)時,就在與抗體的結合維持樹脂-金屬複合體100的分散與抗體的活性的狀態下,使樹脂-金屬複合體100與抗體均勻地接觸的觀點而言,第1pH條件優選為pH為2~7的範圍內的條件,進而更優選為酸性條件,例如pH為2.5~5.5的範圍內。當金屬粒子20為金粒子時,若使樹脂-金屬複合體100與抗體結合時的條件為pH未滿2,則存在因強酸性而導致抗體變質並失活的情況,若pH超過7,則當將樹脂-金屬複合體100與抗體混合時凝聚而難以分散。但是,當不會因強酸性而導致抗體失活時,即便pH未滿2,也可進行處理。另外,當樹脂-金屬複合體100的金屬粒子20為金以外的粒子,例如鈀粒子、或這些的合金等時,就在與抗體的結合維持樹脂-金屬複合體100的分散與抗體的活性的狀態下,使樹脂-金屬複合體100與抗體均勻地接觸的觀點而言,第1pH條件優選為pH為2~10的範圍內的條件,更優選為例如pH為5~9的範圍內。當金屬粒子20為金以外的粒子時,若使樹脂-金屬複合體100與抗體結合時的條件為pH未滿2,則存在因強酸性而導致抗體變質並失活的情況,若pH超過10,則當將樹脂-金屬複合體100與抗體混合時凝聚而難以分散。但是,當不會因強酸性而導致抗體失活時,即便pH未滿2,也可進行處理。步驟A優選為在調整成第1pH條件的結合用緩衝液(BindingBuffer)中進行。例如,在調整成所述pH的結合用緩衝液中混合規定量的樹脂-金屬複合體100,並充分地進行混合。作為結合用緩衝液,例如可使用調整成規定濃度的硼酸溶液等。結合用緩衝液的pH的調整例如可使用鹽酸、氫氧化鈉等來進行。繼而,向所獲得的混合液中添加規定量的抗體,並充分地進行攪拌、混合,由此可獲得標記抗體含有液。以所述方式獲得的標記抗體含有液例如可利用離心分離等固液分離方法,僅分離取出作為固體部分的標記抗體150。[步驟B]在步驟B中,在第2pH條件下對步驟A中所獲得的標記抗體150進行處理,由此進行抑制對於標記抗體150的非特異的吸附的阻隔。在此情況下,在第2pH條件下,使利用固液分離方法而分離取出的標記抗體150分散於液相中。所述阻隔的條件根據樹脂-金屬複合體100中的金屬粒子20的金屬種類而不同。當樹脂-金屬複合體100的金屬粒子20為金粒子時,就保持抗體的活性且抑制標記抗體150的凝聚的觀點而言,第2pH條件例如優選為pH為2~9的範圍內,進而,就抑制標記抗體150的非特異的吸附的觀點而言,更優選為酸性條件,例如pH為2~6的範圍內。若阻隔的條件為pH未滿2,則存在因強酸性而導致抗體變質並失活的情況,若pH超過9,則標記抗體150凝聚而難以分散。另外,當樹脂-金屬複合體100的金屬粒子20為金以外的粒子時,就保持抗體的活性且抑制標記抗體150的凝聚的觀點而言,第2pH條件例如優選為pH為2~10的範圍內,就抑制標記抗體150的非特異的吸附的觀點而言,更優選為pH為5~9的範圍內。若阻隔的條件為pH未滿2,則存在因強酸性而導致抗體變質並失活的情況,若pH超過10,則標記抗體150凝聚而難以分散。步驟B優選為使用調整成第2pH條件的阻隔用緩衝液(BlockingBuffer)來進行。例如,向規定量的標記抗體150中添加調整成所述pH的阻隔用緩衝液,並使標記抗體150均勻地分散於阻隔用緩衝液中。作為阻隔用緩衝液,例如優選為使用不與被檢測物結合的蛋白質的溶液。作為可用於阻隔用緩衝液的蛋白質,例如可列舉:牛血清白蛋白、卵白蛋白、酪蛋白、明膠等。更具體而言,優選為使用調整成規定濃度的牛血清白蛋白溶液等。阻隔用緩衝液的pH的調整例如可使用鹽酸、氫氧化鈉等來進行。標記抗體150的分散例如優選為使用超聲波處理等分散方法。以所述方式獲得均勻分散有標記抗體150的分散液。以所述方式,可獲得標記抗體150的分散液。自所述分散液中,例如可利用離心分離等固液分離方法,僅分離取出作為固體部分的標記抗體150。另外,視需要可實施清洗處理、保存處理等。以下,對清洗處理、保存處理進行說明。(清洗處理)清洗處理是向利用固液分離方法而分離取出的標記抗體150中添加清洗用緩衝液,並使標記抗體150均勻地分散於清洗用緩衝液中。分散例如優選為使用超聲波處理等分散方法。作為清洗用緩衝液,並無特別限定,例如可使用調整成pH為8~9的範圍內的規定濃度的三羥甲基氨基甲烷緩衝液(Trisbuffer)、甘胺醯胺緩衝液(Glycineamidebuffer)、精胺酸緩衝液(Argininebuffer)等。清洗用緩衝液的pH的調整例如可使用鹽酸、氫氧化鈉等來進行。標記抗體150的清洗處理視需要可重複進行多次。(保存處理)保存處理是向利用固液分離方法而分離取出的標記抗體150中添加保存用緩衝液,並使標記抗體150均勻地分散於保存用緩衝液中。分散例如優選為使用超聲波處理等分散方法。作為保存用緩衝液,例如可使用向清洗用緩衝液中添加規定濃度的抗凝聚劑及/或穩定劑而成的溶液等。作為抗凝聚劑,例如可使用:以蔗糖、麥芽糖、乳糖、海藻糖為代表的糖類,或以甘油、聚乙烯醇為代表的多元醇等。作為穩定劑,並無特別限定,例如可使用:牛血清白蛋白、卵白蛋白、酪蛋白、明膠等蛋白質。能夠以所述方式進行標記抗體150的保存處理。在以上的各步驟中,視需要可進而使用表面活性劑,或疊氮化鈉、對羥基苯甲酸酯等防腐劑。[分析物檢測·定量套組]本發明的一實施形態的分析物測定用套組是使用例如側流型色譜用測試條200,根據本實施形態的分析物的測定方法,用以對試樣中所含有的分析物160進行檢測或定量的套組。本實施形態的套組包括:側流型色譜用測試條200,包含薄膜110、及將與所述分析物160特異地結合的捕捉配體固定於薄膜110上而成的判定部130;以及檢測試藥,包含利用具有將多個金屬粒子20固定於樹脂粒子10上而成的結構的樹脂-金屬複合體100對與分析物160特異地結合的抗體進行標記而成的標記抗體150。本實施形態的套組視需要可進而包含其他構成要素。在使用本發明的套組時,可在使試樣中的分析物160與檢測試藥中的標記抗體150接觸來實施步驟(I)後,向測試條200的反應部或試樣添加部120中提供試樣,然後依次實施步驟(II)、步驟(III)。或者,也可將檢測試藥塗布於比測試條200的判定部130更上遊側,適宜進行乾燥而形成反應部後,向所形成的反應部或比所述反應部更上遊側的位置(例如試樣添加部120)添加試樣,然後依次實施步驟(I)~步驟(III)。實施例其次,通過實施例來具體地說明本發明,但本發明不受這些實施例任何限定。在以下的實施例、比較例中,只要事先無特別說明,則各種測定、評價為利用下述方式的。<樹脂-金屬複合體的吸光度測定>關於樹脂-金屬複合體的吸光度,向光學用白板玻璃制單元(光程長度為10mm)中加入製備成0.01wt%的樹脂-金屬複合體分散液(分散介質:水),使用瞬間多重測光系統(multi-meteringsystem)(大冢電子公司製造,MCPD-3700),在金的情況下測定570nm的吸光度。在金的情況下,將570nm中的吸光度為0.9以上設為○(良好),將0.5~未滿0.9設為△(可),將未滿0.5設為×(不可)。<固體成分濃度測定及金屬承載量的測定>向磁製坩堝中加入濃度調整前的分散液1g,並在70℃下進行3小時熱處理。測定熱處理前後的重量,並利用下述式來算出固體成分濃度。固體成分濃度(wt%)=[乾燥後的重量(g)/乾燥前的重量(g)]×100另外,在500℃下,對所述熱處理後的樣品進而進行5小時加熱處理,測定加熱處理前後的重量,並利用下述式來算出金屬承載量。金屬承載量(wt%)=[500℃加熱處理後的重量(g)/500℃加熱處理前的重量(g)]×100<樹脂粒子及樹脂-金屬複合體的平均粒徑的測定>使用圓盤離心式粒度分布測定裝置(CPS圓盤式離心機(DiscCentrifuge)DC24000UHR,CPS儀器有限(CPSinstruments,Inc.)公司製造)進行測定。測定是在使樹脂-金屬複合體分散於水中的狀態下進行。<利用免疫色譜法的評價>使用各實施例等中所製作的樹脂-金屬複合體標記抗體分散液,進行下述所示的利用免疫色譜法的測定,並評價樹脂-金屬複合體分散液的性能。(評價方法)評價是使用流感A型評價用單色屏幕(愛德泰克(Adtec)公司製造),比較5分鐘後、10分鐘後、15分鐘後的顯色水平。在性能評價中,抗原使用流感A型陽性對照(APC)的2倍稀釋列(1倍~1024倍)(APC稀釋前的病毒的濃度為5000FFU/ml)。(評價程序)向96孔盤的各孔中各加入3μl樹脂-金屬複合體標記抗體分散液,並混合APC的2倍稀釋列(1倍~1024倍)及陰性對照各100μl。繼而,向流感A型評價用單色屏幕中添加50μl,並評價5分鐘後、10分鐘後、15分鐘後的顯色水平。顯色水平使用膠體金判定用顏色樣本(愛德泰克公司製造)進行判定。<金屬粒子的平均粒徑的測定>金屬粒子的平均粒徑的測定是根據利用場發射掃描型電子顯微鏡(STEM;日立先端科技(HitachiHigh-Technologies)公司製造,SU-9000)觀測朝帶有碳支撐膜的金屬性網眼滴加樹脂-金屬複合體分散液所製作的基板的圖像,測定金屬粒子的面積平均徑。[實施例1]<樹脂粒子的合成>使愛麗庫特(Aliquat)336[奧德裡奇(Aldrich)公司製造](1.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,2.00g)溶解於80g的純水中後,添加2-乙烯基吡啶(2-VP,9.90g)及二乙烯基苯(DVB,0.100g),在氮氣氣流下,以250rpm、60℃攪拌30分鐘。攪拌後,歷時5分鐘滴加溶解於9.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.100g),以250rpm、60℃攪拌6小時,由此獲得平均粒徑為0.45μm的樹脂粒子。利用離心分離(9000rpm,10分鐘)來使所述樹脂粒子沉澱,去除上清液後,再次分散於純水中,而獲得10wt%的樹脂粒子分散液。<樹脂-金屬複合體的合成>向所述樹脂粒子分散液(3.05g)中添加30mM氯金酸水溶液(80g),並在室溫下放置24小時。其後,利用離心分離(3000rpm,10分鐘)來使樹脂粒子沉澱,並去除上清液,由此去除多餘的氯金酸後,再次分散於55g的純水中,而製備金離子吸附樹脂粒子分散液。歷時8分鐘將所述金離子吸附樹脂粒子分散液45g)滴加至10mM的二甲基胺硼烷水溶液(450ml)中後,在室溫下攪拌2小時,由此獲得平均粒徑為0.6μm的樹脂-金複合體。利用離心分離(3000rpm,120分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於37g的純水中,並利用超濾膜來進行精製,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所述樹脂-金複合體分散液中的樹脂-金複合體的吸光度的結果為1.20。另外,樹脂-金複合體中的金粒子的平均粒徑為22.0nm,金的承載量為49.4wt%。將所製作的樹脂-金複合體的掃描型電子顯微鏡(SEM)照片示於圖3。[實施例2]除向實施例1中所獲得的樹脂粒子分散液(3.05g)中添加10mM的氯金酸水溶液(56g)以外,以與實施例1相同的方法獲得金離子吸附樹脂粒子分散液、平均粒徑為0.6μm的樹脂-金複合體及1wt%的樹脂-金複合體分散液。所述樹脂-金複合體分散液中的樹脂-金複合體的吸光度為1.04。另外,樹脂-金複合體中的金粒子的平均粒徑為7.61nm,金的承載量為36.8wt%。[實施例3]<樹脂粒子的合成>將2-VP(9.90g)及DVB(0.100g)添加至450g的純水中,在氮氣氣流下,以250rpm、60℃攪拌30分鐘。攪拌30分鐘後,歷時5分鐘滴加溶解於9.00g的純水中的AIBA(0.100g),以250rpm攪拌6小時,由此獲得平均粒徑為0.10μm的樹脂粒子。利用離心分離(9000rpm,20分鐘)來使所述樹脂粒子沉澱,去除上清液後,再次分散於純水中,而獲得10wt%的樹脂粒子分散液。<樹脂-金屬複合體的合成>向樹脂粒子分散液(5.0g)中添加30mM的氯金酸水溶液(198g),並在室溫下放置24小時。其後,利用離心分離(3000rpm,10分鐘)來使樹脂粒子沉澱,並去除上清液,由此去除多餘的氯金酸後,再次分散於1.5g的純水中,而製備金離子吸附樹脂粒子分散液。歷時2分鐘將所述金離子吸附樹脂粒子分散液(1.5g)滴加至10mM的二甲基胺硼烷水溶液(65ml)中後,在室溫下攪拌2小時,由此獲得平均粒徑為0.22μm的樹脂-金複合體。向所述樹脂-金複合體中添加10wt%的分散劑(BYK194)600μl並攪拌1小時後,利用離心分離(9000rpm,10分鐘)來進行沉澱,並去除上清液。其後,添加適量的純水再次進行分散,並利用超濾膜來進行精製,而獲得1wt%的樹脂-金複合體分散液。所述樹脂-金複合體分散液中的樹脂-金複合體的吸光度為1.12。另外,樹脂-金複合體中的金粒子的平均粒徑為22.6nm,金的承載量為37.0wt%。[比較例1]<免疫色譜的評價>在著色乳膠(默克密理博(MerckMillipore)公司製造,著色愛斯塔波(Estapor)功能性粒子,K1030,平均粒徑為:392nm,570nm中的吸光度為0.83,400nm中的吸光度為1.11)1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使著色乳膠與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔著色乳膠。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作著色乳膠標記抗體。使用所製作的著色乳膠標記抗體,進行下述所示的利用免疫色譜法的測定,並評價著色乳膠的性能。(評價方法)評價是使用流感A型評價用單色屏幕(愛德泰克公司製造),比較5分鐘後、10分鐘後、15分鐘後的顯色水平。在性能評價中,抗原使用流感A型陽性對照(APC)的2倍稀釋列(1倍~1024倍)(APC稀釋前的病毒的濃度為5000FFU/ml)。(評價程序)向96孔盤的各孔中各加入3μl著色乳膠標記抗體,並混合APC的2倍稀釋列(1倍~1024倍)及陰性對照各100μl。繼而,向流感A型評價用單色屏幕中添加50μl,並評價5分鐘後、10分鐘後、15分鐘後的顯色水平。以下表示其結果。根據所述表1A,確認著色乳膠標記抗體對於16倍稀釋的抗原顯示出良好的顯色。將以上的實施例及比較例的吸光度的結果匯總並示於表1B中。[表1B]實施例1實施例2實施例3比較例1570nm中的吸光度1.201.041.120.83評價○○○△[比較例2]<膠體金的合成>向500ml三口圓底燒瓶中加入1mM氯金酸水溶液250ml,使用加熱回流裝置,一面激烈地攪拌一面使其沸騰,沸騰後添加38.8mM檸檬酸鈉水溶液25ml,並確認溶液自淡黃色變化成深紅色。一面進行攪拌一面繼續加熱10分鐘後,在室溫下進行30分鐘左右的攪拌並放置冷卻。使用孔徑為2μm的薄膜過濾器對溶液進行過濾,移至三角燒瓶中並在陰涼處保存。所製作的粒子的平均粒徑為12.3nm。<免疫色譜的評價>在所獲得的膠體金1ml(OD=10)中混合流感抗體100μg,並在室溫下攪拌約3小時來使膠體金與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔膠體金表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作膠體金標記抗體。使用所製作的膠體金標記抗體,進行下述所示的利用免疫色譜法的測定,並評價膠體金的性能。(評價方法)評價是使用流感A型評價用單色屏幕(愛德泰克公司製造),比較5分鐘後、10分鐘後、15分鐘後的顯色水平。在性能評價中,抗原使用流感A型陽性對照(APC)的2倍稀釋列(1倍~1024倍)(APC稀釋前的病毒的濃度為5000FFU/ml)。(評價程序)向96孔盤的各孔中各加入3μl膠體金標記抗體,並混合APC的2倍稀釋列(1倍~1024倍)及陰性對照各100μl。繼而,向流感A型評價用單色屏幕中添加50μl,並評價5分鐘後、10分鐘後、15分鐘後的顯色水平。以下表示其結果。根據所述表2,確認膠體金標記抗體對於32倍稀釋的抗原顯示出良好的顯色。[比較例3]<樹脂粒子的合成>使愛麗庫特(Aliquat)336[奧德裡奇公司製造](5.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,10.00g)溶解於389.5g的純水中後,添加2-乙烯基吡啶(2-VP,48.00g)及二乙烯基苯(DVB,2.00g),在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在60℃下攪拌30分鐘。攪拌後,歷時2分鐘滴加溶解於50.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.500g),以150rpm、60℃攪拌3.5小時,由此獲得平均粒徑為200nm的樹脂粒子。利用離心分離(9000rpm,60分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(beads)(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、20℃進行攪拌,一面歷時4分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)與528mM的硼酸水溶液(10ml)的混合溶液後,在室溫下攪拌2小時,由此獲得平均粒徑為250nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為1.69。另外,所形成的金粒子的平均粒徑為75.0nm,金的承載量為52.3wt%。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000pm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合體標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表3,確認樹脂-金複合體標記抗體對於16倍稀釋的抗原顯示出良好的顯色。[實施例4]使愛麗庫特(Aliquat)336[奧德裡奇公司製造](1.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,10.00g)溶解於300g的純水中後,添加2-乙烯基吡啶(2-VP,48.00g)及二乙烯基苯(DVB,2.00g),在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在60℃下攪拌30分鐘。攪拌後,歷時0.5分鐘滴加溶解於18.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.500g),以150rpm、60℃攪拌3.5小時,由此獲得平均粒徑為500nm的樹脂粒子。利用離心分離(9000rpm,40分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、20℃進行攪拌,一面歷時2分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)後,在室溫下攪拌2小時,由此獲得平均粒徑為510nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為1.01。另外,所形成的金粒子的平均粒徑為46.3nm,金的承載量為54.2wt%。在所述樹脂-金複合體中,金粒子包括完全地內包於樹脂粒子中的內包金粒子、具有包埋於樹脂粒子內的部位及朝樹脂粒子外露出的部位的部分露出金粒子、及吸附於樹脂粒子的表面的表面吸附金粒子,且至少一部分的金粒子三維地分布於樹脂粒子的表層部。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表4,確認樹脂-金複合體標記抗體對於256倍稀釋的抗原顯示出良好的顯色。[實施例5]使愛麗庫特(Aliquat)336[奧德裡奇公司製造](0.50g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,10.00g)溶解於300g的純水中後,添加2-乙烯基吡啶(2-VP,48.00g)及二乙烯基苯(DVB,2.00g),在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在60℃下攪拌30分鐘。攪拌後,歷時0.5分鐘滴加溶解於18.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.500g),以150rpm、60℃攪拌3.5小時,由此獲得平均粒徑為613nm的樹脂粒子。利用離心分離(9000rpm,40分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、3℃進行攪拌,一面歷時2分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)後,在室溫下攪拌2小時,由此獲得平均粒徑為625nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為0.98。另外,所形成的金粒子的平均粒徑為25.0nm,金的承載量為55.3wt%。在所述樹脂-金複合體中,金粒子包括完全地內包於樹脂粒子中的內包金粒子、具有包埋於樹脂粒子內的部位及朝樹脂粒子外露出的部位的部分露出金粒子、及吸附於樹脂粒子的表面的表面吸附金粒子,且至少一部分的金粒子三維地分布於樹脂粒子的表層部。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表5,確認樹脂-金複合體標記抗體對於256倍稀釋的抗原顯示出良好的顯色。[實施例6]使愛麗庫特(Aliquat)336[奧德裡奇公司製造](1.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,10.00g)溶解於300g的純水中後,添加4-乙烯基吡啶(4-VP,48.00g)及二乙烯基苯(DVB,2.00g),在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在60℃下攪拌30分鐘。攪拌後,歷時2分鐘滴加溶解於18.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.500g),以150rpm、60℃攪拌3.5小時,由此獲得平均粒徑為438nm的樹脂粒子。利用離心分離(9000rpm,45分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、3℃進行攪拌,一面歷時2分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)後,在室溫下攪拌2小時,由此獲得平均粒徑為448nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為0.99。另外,所形成的金粒子的平均粒徑為24.0nm,金的承載量為55.7wt%。在所述樹脂-金複合體中,金粒子包括完全地內包於樹脂粒子中的內包金粒子、具有包埋於樹脂粒子內的部位及朝樹脂粒子外露出的部位的部分露出金粒子、及吸附於樹脂粒子的表面的表面吸附金粒子,且至少一部分的金粒子三維地分布於樹脂粒子的表層部。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表6,確認樹脂-金複合體標記抗體對於256倍稀釋的抗原顯示出良好的顯色。[實施例7]使愛麗庫特(Aliquat)336[奧德裡奇公司製造](1.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,10.00g)溶解於300g的純水中後,添加3-乙烯基吡啶(3-VP,48.00g)及二乙烯基苯(DVB,2.00g),在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在60℃下攪拌30分鐘。攪拌後,歷時2分鐘滴加溶解於18.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.500g),以150rpm、60℃攪拌3.5小時,由此獲得平均粒徑為429nm的樹脂粒子。利用離心分離(9000rpm,45分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、3℃進行攪拌,一面歷時2分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)後,在室溫下攪拌2小時,由此獲得平均粒徑為436nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為1.03。另外,所形成的金粒子的平均粒徑為24.3nm,金的承載量為55.5wt%。在所述樹脂-金複合體中,金粒子包括完全地內包於樹脂粒子中的內包金粒子、具有包埋於樹脂粒子內的部位及朝樹脂粒子外露出的部位的部分露出金粒子、及吸附於樹脂粒子的表面的表面吸附金粒子,且至少一部分的金粒子三維地分布於樹脂粒子的表層部。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表7,確認樹脂-金複合體標記抗體對於256倍稀釋的抗原顯示出良好的顯色。[實施例8]使甲基丙烯酸2-(二異丙基氨基)乙酯(DPA,10.3g)、聚(丙二醇)二丙烯酸酯(0.2g)與聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,2.0g)溶解於85g的純水中後,在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在70℃下攪拌30分鐘。攪拌後,歷時2分鐘滴加溶解於2.00g的純水中的過氧二硫酸銨(ASP,0.10g),以150rpm、70℃攪拌3.5小時,由此獲得平均粒徑為338nm的樹脂粒子。利用離心分離(9000rpm,45分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、3℃進行攪拌,一面歷時2分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)後,在室溫下攪拌2小時,由此獲得平均粒徑為345nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為0.96。另外,所形成的金粒子的平均粒徑為24.6nm,金的承載量為48.5wt%。在所述樹脂-金複合體中,金粒子包括完全地內包於樹脂粒子中的內包金粒子、具有包埋於樹脂粒子內的部位及朝樹脂粒子外露出的部位的部分露出金粒子、及吸附於樹脂粒子的表面的表面吸附金粒子,且至少一部分的金粒子三維地分布於樹脂粒子的表層部。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表8,確認樹脂-金複合體標記抗體對於256倍稀釋的抗原顯示出良好的顯色。[實施例9]<樹脂粒子的合成>使愛麗庫特(Aliquat)336[奧德裡奇公司製造](3.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,10.00g)溶解於300g的純水中後,添加2-乙烯基吡啶(2-VP,49.50g)及二乙烯基苯(DVB,0.50g),在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在60℃下攪拌30分鐘。攪拌後,歷時2分鐘滴加溶解於18.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.250g),以150rpm、60℃攪拌3.5小時,由此獲得平均粒徑為370nm的樹脂粒子。利用離心分離(9000rpm,60分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、20℃進行攪拌,一面歷時2分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)後,在室溫下攪拌2小時,由此獲得平均粒徑為393nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為0.92。另外,所形成的金粒子的平均粒徑為14.9nm,金的承載量為55.8wt%。將所獲得的樹脂-金複合體的表面的掃描型電子顯微鏡(SEM)照片示於圖4,將其剖面的掃描型透射電子顯微鏡(STEM)照片示於圖5。在所述樹脂-金複合體中,金粒子包括完全地內包於樹脂粒子中的內包金粒子、具有包埋於樹脂粒子內的部位及朝樹脂粒子外露出的部位的部分露出金粒子、及吸附於樹脂粒子的表面的表面吸附金粒子,且至少一部分的金粒子三維地分布於樹脂粒子的表層部。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合體標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表9,確認樹脂-金複合體標記抗體對於64倍稀釋的抗原顯示出良好的顯色。[實施例10]<樹脂粒子的合成>使愛麗庫特(Aliquat)336[奧德裡奇公司製造](2.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,10.00g)溶解於300g的純水中後,添加2-乙烯基吡啶(2-VP,48.00g)及二乙烯基苯(DVB,2.00g),在氮氣氣流下,以150rpm、30℃攪拌50分鐘,繼而在60℃下攪拌30分鐘。攪拌後,歷時2分鐘滴加溶解於18.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.500g),以150rpm、60℃攪拌3.5小時,由此獲得平均粒徑為380nm的樹脂粒子。利用離心分離(9000rpm,60分鐘)來進行沉澱,去除上清液後,再次分散於純水中,將所述操作進行3次後,利用透析處理來去除雜質。其後,進行濃度調整而獲得10wt%的樹脂粒子分散液。向所述樹脂粒(50ml)中添加純水1233ml後,添加30mM氯金酸水溶液(100ml),並在室溫下放置24小時。其後,利用離心分離(3100rpm,30分鐘)來使樹脂粒子沉澱,並去除上清液,將所述作業重複3次,由此去除多餘的氯金酸。其後,進行濃度調整,而製備2.5wt%金離子吸附樹脂粒子分散液。繼而,向純水1580ml中添加所述2.5wt%金離子吸附樹脂粒子分散液(42.4ml),一面以160rpm、20℃進行攪拌,一面歷時2分鐘滴加528mM的二甲基胺硼烷水溶液(10ml)後,在室溫下攪拌2小時,由此獲得平均粒徑為399nm的樹脂-金複合體。利用離心分離(3100rpm,60分鐘)來使所述樹脂-金複合體沉澱,去除上清液後,再次分散於純水中,將所述作業重複3次後,利用透析處理來進行精製、濃度調整,由此獲得1wt%的樹脂-金複合體分散液。根據所述方法測定所製作的樹脂-金複合體的吸光度的結果為0.96。另外,所形成的金粒子的平均粒徑為25.0nm,金的承載量為53.2wt%。將所獲得的樹脂-金複合體的表面的掃描型電子顯微鏡(SEM)照片示於圖6,將其剖面的掃描型透射電子顯微鏡(STEM)照片示於圖7。在所述樹脂-金複合體中,金粒子包括完全地內包於樹脂粒子中的內包金粒子、具有包埋於樹脂粒子內的部位及朝樹脂粒子外露出的部位的部分露出金粒子、及吸附於樹脂粒子的表面的表面吸附金粒子,且至少一部分的金粒子三維地分布於樹脂粒子的表層部。<免疫色譜的評價>在所獲得的樹脂-金複合體分散液1ml(0.1wt%)中混合流感抗體100μg,並在室溫下攪拌約3小時來使樹脂-金複合體與抗體結合。以最終濃度變成1%的方式添加牛血清白蛋白溶液,並在室溫下攪拌2小時來阻隔樹脂-金複合體表面。以12000rpm、4℃進行5分鐘離心分離後回收,並懸浮於含有0.2%牛血清白蛋白的緩衝液中來製作樹脂-金複合體標記抗體分散液。使用所製作的樹脂-金複合體標記抗體分散液,進行利用免疫色譜法的測定,並評價所述樹脂-金複合體分散液的性能。以下表示其結果。根據所述表10,確認樹脂-金複合體標記抗體對於256倍稀釋的抗原顯示出良好的顯色。若比較實施例9的圖5與實施例10的圖7的剖面圖像,則金粒子的60%~100%、優選為75%~100%存在於自樹脂粒子的表面起在深度方向上為粒子半徑的50%的範圍內的實施例10的免疫色譜的檢測感度更優異。[關於標記抗體的製作的試驗例][製作例1]<樹脂粒子的合成>使愛麗庫特(Aliquat)336[奧德裡奇公司製造](1.00g)及聚乙二醇甲基乙基醚甲基丙烯酸酯(PEGMA,2.00g)溶解於80g的純水中後,添加2-乙烯基吡啶(2-VP,9.90g)及二乙烯基苯(DVB,0.100g),在氮氣氣流下,以250rpm、60℃攪拌30分鐘。攪拌後,歷時5分鐘滴加溶解於9.00g的純水中的2,2-偶氮雙(2-甲基丙脒)二鹽酸鹽(AIBA,0.100g),以250rpm、60℃攪拌6小時,由此獲得平均粒徑為0.36μm的樹脂粒子A-1。利用離心分離(9000rpm,10分鐘)來使所述A-1進行沉澱,去除上清液後,再次分散於純水中,而獲得2.1wt%的樹脂粒子分散液B-1。<樹脂-金複合體的合成>向所述B-1(19.09g)中添加30mM氯金酸水溶液(106.6g),並在室溫下放置24小時。其後,利用離心分離(3000rpm,10分鐘)來使樹脂粒子沉澱,並去除上清液,由此去除多餘的氯金酸後,再次分散於40g的純水中,而製備金離子吸附樹脂粒子分散液C-1。歷時4分鐘將所述C-1(20g)滴加至3.3mM的二甲基胺硼烷水溶液(600ml)中後,在8℃下攪拌1小時,進而在室溫下攪拌5小時,由此獲得平均粒徑為0.38μm的樹脂-金複合體D-1。利用離心分離(3000rpm,120分鐘)來使所述D-1沉澱,去除上清液後,添加適量的純水來再次進行分散,然後利用超濾膜來進行精製,由此獲得1wt%的樹脂-金複合體分散液E-1。根據所述方法測定E-1中的樹脂-金複合體F-1的吸光度的結果為1.0。另外,F-1中的金粒子的平均粒徑為22.0nm,金的承載量為49.1wt%。[試藥等]在試驗例、參考試驗例中使用以下的試藥等。抗流感A型單株抗體(7.15mg/mL/PBS):愛德泰克股份有限公司製造。結合用緩衝液a:100mM利用HCl將硼酸溶液調整成pH≒3。結合用緩衝液b:100mM利用HCl將硼酸溶液調整成pH≒4。結合用緩衝液c:100mM利用HCl將硼酸溶液調整成pH≒5。結合用緩衝液d:100mM硼酸溶液pH≒6.5。結合用緩衝液e:100mM利用NaCl將硼酸溶液調整成pH≒7.5。結合用緩衝液f:100mM利用NaCl將硼酸溶液調整成pH≒8.5。結合用緩衝液g:50mM2-嗎啉基乙磺酸溶液pH≒3.8。阻隔用緩衝液a:利用HCl將1wt%牛血清白蛋白溶液調整成pH≒5。阻隔用緩衝液b:利用HCl將1wt%牛血清白蛋白溶液調整成pH≒7。阻隔用緩衝液c:利用HCl將1wt%牛血清白蛋白溶液調整成pH≒8.5。阻隔用緩衝液d:利用HCl將1wt%牛血清白蛋白溶液調整成pH≒9.5。清洗用緩衝液:利用HCl將5mM三羥甲基氨基甲烷溶液調整成pH≒8.5。保存用緩衝液:以變成10wt%濃度的方式將蔗糖添加至清洗用緩衝液中。流感A型陽性對照(APC):使用檢體處理液(愛德泰克股份有限公司製造)對流感A型病毒鈍化抗原(愛德泰克股份有限公司製造)進行100倍稀釋來製備。APC的抗原濃度相當於5000FFU/ml。陰性對照:檢體處理液(愛德泰克股份有限公司製造)。AuNCP粒:製作例1中所獲得的樹脂-金複合體(1wt%;平均粒徑為380nm)。[試驗例1](結合步驟)向微型管[IBIS(註冊商標;亞速旺(ASONE)公司製造)2mL]中投入作為樹脂-金屬複合體的AuNCP粒0.1mL,並添加0.9mL的結合用緩衝液a。利用顛倒混合而充分地混合後,添加抗流感A型單株抗體100μg,並在室溫下歷時3小時進行顛倒攪拌,而獲得含有利用樹脂-金屬複合體進行了標記的抗流感A型單株抗體的標記抗體含有液A-1。(阻隔步驟)繼而,對標記抗體含有液A-1進行冰浴冷卻後,歷時5分鐘以12000rpm進行離心分離,去除上清液後,向固體成分殘渣中添加1mL的阻隔用緩衝液a,歷時10秒~20秒進行超聲波分散處理,進而,在室溫下歷時2小時進行顛倒攪拌,而獲得標記抗體含有液B-1。(清洗處理)繼而,對標記抗體含有液B-1進行冰浴冷卻後,歷時5分鐘以12000rpm進行離心分離,去除上清液後,向固體成分殘渣中添加清洗用緩衝液1mL,歷時10秒~20秒進行超聲波分散處理。將所述操作重複3次來作為清洗處理。(保存處理)繼而,在冰浴冷卻後,歷時5分鐘以12000rpm進行離心分離,去除上清液後,向固體成分殘渣中添加保存用緩衝液1mL,歷時10秒~20秒進行超聲波分散處理,由此獲得標記抗體含有液C-1。[試驗例2]在試驗例1的結合步驟中使用結合用緩衝液b來代替結合用緩衝液a,除此以外,以與試驗例1相同的方式獲得標記抗體含有液A-2、標記抗體含有液B-2、標記抗體含有液C-2。[試驗例3]在試驗例1的結合步驟中使用結合用緩衝液c來代替結合用緩衝液a,除此以外,以與試驗例1相同的方式獲得標記抗體含有液A-3、標記抗體含有液B-3、標記抗體含有液C-3。[試驗例4]在試驗例1的結合步驟中使用結合用緩衝液d來代替結合用緩衝液a,除此以外,以與試驗例1相同的方式獲得標記抗體含有液A-4、標記抗體含有液B-4、標記抗體含有液C-4。[參考試驗例1]當在試驗例1的結合步驟中使用結合用緩衝液e來代替結合用緩衝液a時,樹脂-金屬複合體凝聚,因此難以獲得標記抗體含有液。[參考試驗例2]當在試驗例1的結合步驟中使用結合用緩衝液f來代替結合用緩衝液a時,樹脂-金屬複合體凝聚,因此難以獲得標記抗體含有液。[試驗例5]在試驗例1的阻隔步驟中使用阻隔用緩衝液b來代替阻隔用緩衝液a,除此以外,以與試驗例1相同的方式獲得標記抗體含有液B-5、標記抗體含有液C-5。[試驗例6]在試驗例1的阻隔步驟中使用阻隔用緩衝液c來代替阻隔用緩衝液a,除此以外,以與試驗例1相同的方式獲得標記抗體含有液B-6、標記抗體含有液C-6。[參考試驗例3]在試驗例1的阻隔步驟中使用阻隔用緩衝液d來代替阻隔用緩衝液a的結果,結合步驟後的標記抗體顯示出良好的分散性,但在阻隔步驟後標記抗體凝聚,難以獲得標記抗體含有液。[試驗例7]在試驗例1的結合步驟中使用結合用緩衝液g來代替結合用緩衝液a,除此以外,以與試驗例1相同的方式獲得標記抗體含有液A-7、標記抗體含有液B-7、標記抗體含有液C-7。<評價方法>評價是使用流感A型評價用單色屏幕(愛德泰克公司製造),比較5分鐘後、10分鐘後、15分鐘後的顯色水平。顯色水平使用膠體金判定用顏色樣本(愛德泰克公司製造)進行判定。在篩選評價中,抗原使用流感A型陽性對照(APC)。在性能評價中,抗原使用APC的2倍稀釋列(1倍~1024倍稀釋)。<篩選評價>向96孔盤的7個孔中各加入3μL試驗例1~試驗例7中所獲得的標記抗體含有液C-1~標記抗體含有液C-7,並在各孔中混合APC100μL。繼而,向流感A型評價用單色屏幕中各添加50μL,並評價5分鐘後、10分鐘後、15分鐘後的顯色水平。將其結果示於表11。再者,表11中的數值越大,表示顯色水平越高(顯色越強)。[表11]根據表11,確認試驗例1中所獲得的抗體標記含有液C-1顯示出最強的顯色,具有優異的標記性能。<性能評價>向96孔盤的12個孔中各加入3μL試驗例1中所獲得的標記抗體含有液C-1,並混合APC的2倍稀釋列(1倍~1024倍稀釋,分別表示為APC×1~APC×1024)及陰性對照各100μL。繼而,向流感A型評價用單色屏幕中添加50μL,並評價5分鐘後、10分鐘後、15分鐘後的顯色水平。將其結果示於表12。再者,表12中的數值越大,表示顯色水平越高(顯色越強)。根據表12,確認試驗例1中所獲得的標記抗體含有液C-1對於256倍稀釋的抗原也顯示出良好的顯色,具有優異的標記性能。以上,以例示的目的對本發明的實施形態進行了詳細說明,但本發明並不受所述實施形態限制。本國際申請主張基於2014年7月1日所申請的日本專利特願2014-136356號、及2014年7月1日所申請的日本專利特願2014-136357號的優先權,並將所述申請的所有內容引用於本發明中。[符號的說明]10:樹脂粒子20:金屬粒子30:內包金屬粒子40:部分露出金屬粒子50:表面吸附金屬粒子60:表層部100:樹脂-金屬複合體110:薄膜120:試樣添加部130:判定部131:捕捉配體140:吸液部150:標記抗體160:分析物170:複合體200:測試條當前第1頁1 2 3