用於有機電致發光元件的有機銥配合物的製作方法
2023-10-31 13:44:27 4
本發明涉及一種提供適合用作有機電致發光(electroluminescence,EL)元件用的發光材料的有機銥配合物的技術,特別是涉及一種可作為紅色發光材料使用的有機銥配合物。
背景技術:
目前有機EL元件在下一代顯示器或照明的技術開發方面備受期待。其特徵為:消耗電力少,可薄型化,並具有反應速度優異,以及在暗處與明處均可清晰地顯示影像等優點。
作為該有機EL元件的基本結構,以一對電極夾住一層或多層有機化合物的三明治結構是適用的。具體而言,有人提出以陰極/電子輸送層/發光層/空穴輸送層/陽極/玻璃基板這樣的三明治結構為主要構成,並為了進一步提高特性而適當追加空穴(電子)注入層、緩衝層、層間絕緣膜等的結構的元件。在位於三明治結構中心的發光層中使用各種發光材料,其特性為:尋求使從陰極或陽極輸送的電子或空穴容易流動、發光效率優異、具有耐久性等。
由於這樣的要求特性,相對於以往適用的螢光材料,目前尋求開發磷光材料來作為用於有機EL元件的發光材料。在有機EL元件中,因為激發單重態與激發三重態產生激發分子的機率為1:3,故相對於通過從激發單重態回到基態而發光的螢光材料,通過從激發三重態狀態回到基態而顯示磷光的磷光材料更受到矚目。已經有人開發各種有機金屬配合物作為這種磷光材料,例如,有人提出如下式所示的有機金屬配合物,其為將具有雜環且具備C-N結構的配位基(C-N配位基)與β-二酮等的配位基配位於鉑或銥等金屬原子而成。具體而言,專利文獻1公開了一種包含具備兩個苯環的配位基(二苯甲醯基甲烷)作為β-二酮配位基的有機銥配合物(專利文獻1的S02等)。另外,專利文獻2公開了一種包含具有兩個經丁氧基取代的苯環的配位基(四-丁氧基二苯基二酮)作為β-二酮配位基的有機鉑配合物(專利文獻2的式[1-1])。以上專利文獻所記載的有機金屬配合物中,通過應用具有苯環的配位基作為β-二酮配位基,從而謀求發光效率的提高。
[化1]
[現有技術文獻]
[專利文獻]
專利文獻1:日本特開2005-35902號公報
專利文獻2:日本特開2008-222635號公報
技術實現要素:
發明所要解決的問題
另外,目前已知可用「電流效率(cd/A)」與「量子效率(%)」作為有機EL元件的發光效率的評價基準。電流效率表示相對單位電流量的亮度(或考量光視效能的光強度),另一方面,量子效率為作為光能可取出的光子數相對於消耗功率(注入的載子數)的比率。量子效率可排除在消耗功率當中無法釋出為光能的部分(例如電阻所導致的損耗量)。因此,相較於電流效率,量子效率可以說是能夠接近有機EL元件的實際發光效率的評價。在此背景下,若從專利文獻1、2來看,雖然是針對作為發光效率的電流效率高的有機金屬配合物來進行研究,但其並非一定具有高量子效率。另外,一般而言,特別是呈現紅色的有機金屬配合物相較於藍色或綠色等,難以呈現高量子效率。這是由於被稱為能隙定律的分子光學上的本質特性決定的。因為若電子躍遷的能隙變小,則從激發狀態回到基態時,不會發光的無輻射去活化速度會以指數函數方式增加。另外,為了在長波長側發出紅光而導入複雜的C-N配位基(π共軛系的芳香族)這種結構化學上的重要原因,也可被列舉作為量子效率難以變高的理由。
再者,將以往的有機金屬配合物安裝在有機EL元件時,為了應對延長壽命的要求,而必須提高熱穩定性等的耐久性。在這一點上,專利文獻1、2僅進行上述電流效率和發光亮度的評價,而未對熱穩定性進行具體研究。
於是,本發明的目的在於提供一種有機金屬配合物,其作為有機EL元件用的發光材料,可實現高量子效率的電致發光,特別是提供了針對紅色電致發光而言量子效率高的有機金屬配合物。另外,本發明提供一種耐熱性比以往的配合物高的有機金屬配合物。
用於解決問題的手段
為了解決上述問題,本申請發明人等著眼於具有以銥作為中心原子的有機銥配合物。作為有機金屬配合物,雖然如專利文獻2所述那樣開發出一種鉑配合物,但鉑配合物的平面性高,作為中心元素的鉑原子的配位基具有空位,從而容易發生能量損耗。具體而言,其受到下述各種相互作用的影響:締合(會合)·準分子(エキシマー)形成等的分子間相互作用(所謂的自我組織)、或與溶劑·基質(母材)等介質的相互作用、或更進一步與共存的其他分子的締合等。相對於此,有機銥配合物中的三個配位基形成立體的構象,不會發生上述鉑配合物般的各種相互作用,而不易產生能量損耗,故本發明人認為其容易成為高量子效率的材料。
再者,關於量子效率,著眼於決定量子效率的因素之一的發光材料的「發光(PL)量子產率」。如下式所示,在將量子效率大致分為「外部量子效率」與「內部量子效率」的情況下,此PL量子產率為決定內部量子效率的因素之一。發光材料追求高內部量子效率,作為內部量子效率的決定因素,「激子產生效率」及「PL量子產率」產生的影響特別大。其中「激子產生效率」取決於螢光材料或磷光材料的差別,故為了提高內部量子效率,高PL量子產率變得重要。另外,下式中的載子平衡(キャリアバランス)為由材料的組合或膜厚控制等的元件結構所決定的因素。
[量子效率]
外部量子效率=(光萃取效率)×內部量子效率
內部量子效率=(激子產生效率)×(PL量子產率)×(載子平衡)
由於上述理由,本申請發明人對高PL量子產率的發光性有機銥配合物進行深入研究。結果發現具有經叔丁基取代的苯基作為β-二酮配位基的有機銥配合物,從而完成以下的本發明。
即,本發明涉及一種用於有機電致發光(EL)元件的有機銥配合物,其如下式所示,為將C-N配位基與β-二酮配位基配位於銥原子而形成,其中所述C-N配位基包括雜環與兩個苯環進行縮環而成的三環繫結構的取代基,所述β-二酮配位基由具有兩個經叔丁基取代的苯基的丙-1,3-二酮構成。
[化2]
(上式中,R1、R2、R3為叔丁基或氫原子,且至少具有1個以上的叔丁基;在具有兩個叔丁基的情況下,也可互相鍵合而形成飽和烴環;A為具有含氮雜環的取代基;X為雜原子)。
本發明的主要特徵在於採用經叔丁基取代的苯基這種大型取代基作為β-二酮,在採用該β-二酮的同時,對於C-N配位基也採用特定結構。具體而言,採用包含經雜環與兩個苯環縮環的三環繫結構、以及含氮雜環的配位基來作為C-N配位基。在此方面,以往技術中,在設計有機金屬配合物的結構時,對於C-N配位基,主要考量的是波長位移方面,只要是可呈現目標發光色(紅、藍、綠等)的結構,即可從大量列舉的結構中任意選擇。即,在以往,通過使β-二酮的結構改變,來謀求發光效率的提高。
相對於此,本申請發明人等認為,為了得到穩定且量子效率高的有機銥配合物,不僅要將β-二酮配位基設為特定結構,更需要考量與β-二酮配位基的相容性來設計C-N配位基的結構。然而,在設計有機銥配合物的分子時,由於Ir的自旋與軌道之間的相互作用複雜,從而難以用機械計算等方法算出PL量子產率數值。因此,在研究具體的分子結構時,雖然可以考量能級來研究各配位基的可能,但對於實際上能否成為PL量子產率高的配合物,必須考量合成配合物的可能性,並且確認可實際合成的配合物的實驗結果。接著,以上研究的結果發現,只要是這樣的有機銥配合物,其以具有兩個經叔丁基取代的苯基的丙-1,3-二酮作為β-二酮配位基,並以包含經雜環與兩個苯基縮環的三環繫結構、以及含氮雜環的骨架作為C-N配位基,即可實現高量子效率,進而完成上述本發明。另外,上述有機銥配合物的耐熱性也高於以往的配合物。
以下對本發明的有機銥配合物進行詳細說明。
本發明的有機銥配合物通過將兩個C-N配位基及β-二酮配位於3價的銥原子而得到。兩個C-N配位基為彼此相同的結構,β-二酮配位基具有線對稱的結構。以下說明C-N配位基及β-二酮配位基的具體結構。
在本發明中所應用的β-二酮配位基由如下式所示的具有兩個經叔丁基取代的苯基的丙-1,3-二酮所構成。
[化3]
上式中,R1、R2、R3為叔丁基或氫原子。一個苯基至少具有一個以上的叔丁基,優選具有兩個以上的叔丁基。兩個叔丁基可互相鍵合而形成飽和烴環。
以下顯示特別優選的β-二酮配位基的結構。下式中,t-Bu表示叔丁基。
[化4]
接著說明C-N配位基。本發明所應用的C-N配位基的通式如下式所示。
[化5]
在上述C-N配位基中,上側的取代基為經雜環與兩個苯環縮環的三環繫結構。該三環繫結構中的X為雜原子。X優選為氧原子(O)或硫原子(S)。在此情況下,三環繫結構為二苯並呋喃(氧芴)或二苯並噻吩(硫芴)。
配置於C-N配位基下側的A為具有含氮雜環的取代基。該雜環優選為五元環或六元環。另外,A優選為含氮雜環與苯環進行縮合的結構。A的雜環或苯環可在側鏈上具有任意的取代基,該取代基可為供電子基團或吸電子基團中的任一種。取代基的例子可列舉出:烷基(-R,碳原子數1至5)、烷氧基(-OR,碳原子數1至3)、滷素原子(特別是氟原子)、滷化烷基(碳原子數1至5)等。A的雜環也可進一步具有氮(N)以外的雜原子,氮以外的優選的雜原子為硫原子(S)或氧原子(O)。
取代基A特別優選為下式所示的取代基的任一種。
[化6]
以上所說明的本發明的有機銥配合物具有高發光效率,在高分子薄膜中摻雜4wt%時,PL量子產率ΦPL容易變高(例如變為0.4以上)。另外,本發明的有機銥配合物的分解溫度也高,並且其熱穩定性良好。由以上所述,本發明的有機銥配合物適合作為發光層安裝於有機EL元件。
對於本發明的有機銥配合物,可在通過使銥鹽與構成C-N配位基的含氮化合物進行加熱反應而得到前驅體之後,使該前驅體與β-二酮化合物進行加熱反應來合成。此外,對於本發明的有機銥配合物,也可通過使金屬鹽與β-二酮化合物反應後,再使含氮化合物進行反應而合成。用來得到前驅體的加熱反應優選在80℃至130℃進行12至24小時;與β-二酮的加熱反應優選在60℃至130℃進行0.5至12小時。反應優選在溶劑存在下進行。上述合成反應所使用的銥鹽優選為氯化物(IrCl3)。另外,其使用形態可為氯化物的水合物。
在以上所說明的有機銥配合物應用在有機EL元件的情況下,可通過旋轉塗布法、真空沉積法等方法形成發光層。利用旋轉塗布法來形成元件較簡便且成本較低。
[發明的效果]
本發明的有機銥配合物的PL量子產率及耐熱性高於已知的配合物,適合用作有機EL元件的發光材料。
附圖說明
[圖1]實施方案中的有機銥配合物的熱分解特性結果。
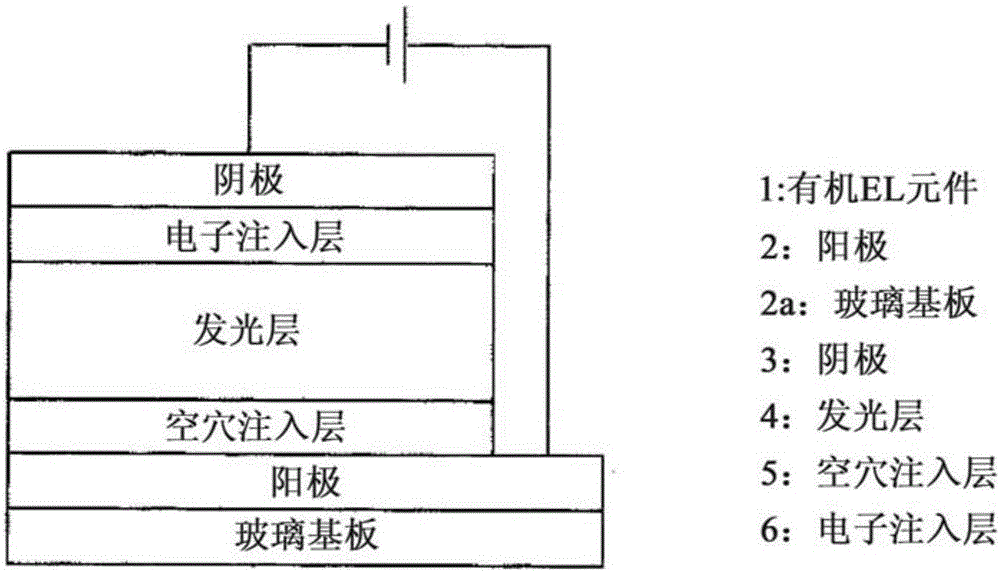
[圖2]實施方案中所製作的有機EL元件的剖面示意圖。
[圖3]將實施方案的有機銥配合物用於發光材料的有機EL元件的電致發光特性評價結果。
具體實施方式
以下說明本發明的最佳實施方案。
合成以下所示的有機銥配合物,並對所得的配合物進行量子效率及熱分解特性的評價。
[化7]
[化8]
關於各銥配合物的合成方法,以配合物1-A的合成方法為例進行概略說明。合成β-二酮化合物(A)及C-N配位基(1:2-(二苯並[b,d]呋喃-4-基)喹啉),並使配位基(1)與氯化銥進行反應從而合成前驅體(1)。接著,通過使前驅體(1)與β-二酮化合物進行反應,得到銥配合物1-A。關於其他配合物,同樣地通過合成β-二酮化合物(B)、配位基(2)至(4)及前驅體(2)至(4),並使各前驅體與各β-二酮化合物進行反應而得到。
初始原料及合成所使用的試劑及溶劑均使用未進行精製的市售試劑等級。對於乾燥四氫呋喃,購入市售的脫水四氫呋喃直接使用。另外,使用關東化學公司制的球狀矽膠(中性),作為柱層析法所使用的填充劑。
使用質子核磁共振(1H NMR)光譜及質量分析(質(MS)譜)來鑑別所合成的化合物。使用Jeol JNM-ECX400分光光度計(400MHz)或Jeol JNM-ECS400分光光度計(400MHz)來測定1H NMR光譜。MS譜則以α-氰基-4-羥基肉桂酸(CHCA)為基質,通過飛行時間(TOF)型質量分析,對以基質輔助雷射解吸離子化法(MALDI法)進行離子化的試樣進行測定(MALDI-TOF-MS譜)。使用Shimadzu-Kratos AXIMA-CFR PLUS TOF Mass質量分析裝置進行測定。對於元素分析,以乙醯苯胺為標準物質,通過「ジェイ·サイエンスラボ」制JM-10元素分析裝置進行測定。
首先說明β-二酮化合物(A)及(B)的合成方法。
β-二酮化合物(A)的合成
合成二丁基苯甲酸甲酯與(二丁基苯甲酸甲酯)乙-1-酮後,通過使用該兩個化合物的合成反應,得到β-二酮化合物(A)。
在氮氣氣氛下,在0℃將濃硫酸(0.9mL)滴至3,5-二-叔丁基苯甲酸(3.00g,12.8mmol)與甲醇(9mL)的混合物中,之後,一邊攪拌一邊使其加熱回流1小時。冷卻後,加入氯仿(100mL),再加入水(100mL)並在分液漏鬥內進行震蕩,從而分離有機層。重複進行一次該操作後,將分離的有機層合併為一。再以飽和碳酸氫鈉水溶液(50mL)及飽和食鹽水(50mL)清洗該有機層後,加入適量無水硫酸鎂以使其乾燥。通過過濾除去硫酸鎂後,以蒸發器餾去溶劑,並在減壓下使殘渣在乾燥器內進行乾燥,由此得到3,5-二-叔丁基苯甲酸甲酯。所得化合物為白色固體,其產率為92%(2.92g,11.8mmol)。以上述方法合成的化合物的特性(1H NMR、TOF MS)如下所示。
1H NMR(CDCl3):δ1.35(s,18H),3.91(s,3H),7.62(t,J=2.0Hz,1H),7.89(d,J=2.0Hz,2H)。
MALDI-TOF MS:m/z 249([M+H]+)。
[化9]
將3,5-二-叔丁基苯甲酸(3.00g,12.8mmol)加入到乾燥四氫呋喃(120mL)中,在氮氣氣氛下攪拌同時進行冷卻,直到變成0℃以下為止。將甲基鋰的3.0M的二乙氧基甲烷溶液(15mL)滴加至該混合物,之後升溫至室溫並攪拌2小時。將6M的鹽酸加入到該反應混合物從而使其成為酸性後,以氯仿(100mL×2)進行萃取。使所得的有機層合併為一,並以水(50mL×2)、飽和碳酸氫鈉水溶液(50mL)及飽和食鹽水(50mL)進行清洗後,加入適量無水硫酸鎂從而進行乾燥。通過過濾除去硫酸鎂後,以蒸發器餾去溶劑,以矽膠柱層析法(展開溶劑:氯仿)對殘渣進行精製,由此得到1-(3,5-二-叔丁基苯基)乙-1-酮。所得的化合物為無色液體,其產率為75%(2.23g,9.60mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3)δ1.37(s,18H),2.60(s,3H),7.64(t,J=1.6Hz,1H),7.80(d,J=1.6Hz,2H)。
MALDI-TOF MS:m/z 232(M+)。
[化10]
將3,5-二-叔丁基苯甲酸甲酯(2.92g,11.8mmol)與氫化鈉(60%油分散;1.27g,31.8mmol)加入到乾燥THF(23mL)中,在氮氣氣氛下,在室溫進行攪拌。在30分鐘之內,向其中滴加使1-(3,5-二-叔丁基苯基)乙-1-酮(2.23g,9.60mmol)溶解於乾燥THF(23mL)的溶液。之後,在60℃下攪拌所得的反應混合物24小時。冷卻後,加入1M的鹽酸使其成為酸性後,以氯仿(100mL×2)進行萃取。將所得的有機層合併為一,並以水(50mL×2)、飽和碳酸氫鈉水溶液(50mL)及飽和食鹽水(50mL)進行清洗後,加入適量無水硫酸鎂從而進行乾燥。通過過濾除去硫酸鎂後,以蒸發器餾去溶劑,以矽膠柱層析法(展開溶劑:氯仿)對殘渣進行精製,由此得到1,3-雙(3,5-二-叔丁基苯基)丙-1,3-二酮(β-二酮A)。所得的化合物為琥珀色的漿狀物質,其產率為49%(2.12g,4.73mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3)δ1.38(s,36H),6.78(s,1H),7.63(t,J=2.0Hz,2H),7.78(d,J=2.0Hz,4H),16.9(brs,1H)。
MALDI-TOF MS:m/z 449([M+H]+)。
[化11]
β-二酮化合物(B)的合成
通過使用1,2,3,4-四氫-1,1,4,4-四甲基萘與丙二醯氯的合成反應,得到β-二酮化合物(B)。
將1,2,3,4-四氫-1,1,4,4-四甲基萘(5.00g,26.6mmol)、丙二醯氯(1.35g,9.58mmol)及氯化鋁(5.51g,41.3mmol)添加到二硫化碳(27mL)中,並在50℃下加熱攪拌3小時。接著,冷卻後,添加冷卻的2mol/L的鹽酸(27mL)並移至分液漏鬥,以氯仿進行萃取。進一步以水清洗有機層,並以蒸發器餾去溶劑後,在殘渣中加入濃鹽酸(3.5mL)與氯仿(35mL),使其加熱回流9小時。冷卻後,將混合物移至分液漏鬥,並以水及飽和食鹽水進行清洗。以無水硫酸鎂使有機層乾燥後,以旋轉蒸發器餾去溶劑。以矽膠柱層析法(展開溶劑;乙酸乙酯:己烷=1:2(v/v))對殘渣進行精製,由此以39%的產率得到β-二酮(B)(1.66g,3.74mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.30(s,12H),1.34(s,12H),1.71(m,8H),6.76(s,1H),7.40(d,J=8.0Hz,2H),7.68(dd,J=8.0及2.0Hz,2H),7.94(d,J=2.0Hz,2H),16.96(brs,1H)。
MALDI-TOF MS:m/z 445([M+H]+)。
[化12]
接著說明C-N配位基(1)至(4)及前驅體(1)至(4)的合成方法。
C-N配位基(1)的合成
依照下式,合成2-(二苯並[b,d]呋喃-4-基)喹啉作為C-N配位基(1)。
[化13]
將二苯並[b,d]呋喃-4-基硼酸(1.44g,6.79mmol)、2-氯喹啉(1.24g,7.58mmol)、四(三苯基膦)鈀(0.658g,0.569mmol)及碳酸鉀(14.4g,104mmol)的混合物添加到1,2-二甲氧基乙烷(75mL)、乙醇(75mL)及水(75mL)的混合溶劑中,在氮氣氣氛下,在設定為100℃的水浴中使其加熱回流18小時。使其冷卻後,以蒸發器餾去有機溶劑,並加入100mL的氯仿。以水(2×100mL)及飽和食鹽水(100mL)清洗該混合物,並加入適量硫酸鎂從而使其乾燥。通過過濾除去硫酸鎂後,以蒸發器餾去濾液的溶劑。以矽膠柱層析法(展開溶劑:氯仿)對得到的殘渣進行精製,並以79%的產率得到2-(二苯並[b,d]呋喃-4-基)喹啉(1.59g,5.38mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ7.39(t,J=7.6Hz,1H),7.48-7.53(m,1H),7.55-7.59(m,2H),7.66(d,J=8.7Hz,1H),7.76(dt,J=1.4及7.6Hz,1H),7.89(d,J=7.6Hz,1H),8.02(d,J=7.6Hz,1H),8.06(dd,J=1.4及7.6Hz,1H),8.23(d,J=8.7Hz,1H),8.34(d,J=8.7Hz,1H),8.43(dd,J=1.4及7.6Hz,1H),8.52(d,J=8.7Hz,1H)。
MALDI-TOF MS:m/z 296([M+H]+)。
前驅體(1)的合成
依照下式,使C-N配位基(1)與氯化銥進行反應從而得到前驅體(1)。在氮氣氣氛下,在油浴中加熱2-(二苯並[b,d]呋喃-4-基)喹啉(3.50g,11.9mmol)與2-乙氧基乙醇(210mL)的混合物。若溶液的溫度達到100℃,則加入氯化銥三水合物(1.60g,4.54mmol)與水(70mL)的混合物,並在120℃下攪拌所得的反應混合物10小時。冷卻後,將水(175mL)加入到反應混合物中,進行過濾並回收沉澱物,並以適量的甲醇進行清洗,由此以83%的產率得到前驅體(1)(3.08g,1.89mmol)。所得的化合物為難溶性固體。不進行進一步精製,而將其用於下述銥配合物的合成。
[化14]
C-N配位基(2)的合成
依照下式,合成1-(二苯並[b,d]呋喃-4-基)異喹啉作為C-N配位基(2)。
[化15]
將二苯並[b,d]呋喃-4-基硼酸(5.00g,23.6mmol)、1-氯異喹啉(4.19g,25.6mmol)、四(三苯基膦)鈀(0)(2.27g,1.96mmol)及碳酸鉀(49.2g,356mmol)的混合物添加到1,2-二甲氧基乙烷(150mL)、乙醇(150mL)及水(150mL)的混合溶劑中,在氮氣氣氛下,使其加熱回流12小時。冷卻後,向反應混合物中添加水與乙酸乙酯並在分液漏鬥內進行震蕩,分離有機層後,以飽和食鹽水進行清洗,並加入適量硫酸鎂從而使其乾燥。通過過濾除去硫酸鎂後,以蒸發器餾去濾液的溶劑。以矽膠柱層析法(展開溶劑;氯仿:己烷=1:3(v/v))對得到的殘渣進行精製,由此以74%的產率得到1-(二苯並[b,d]呋喃-4-基)異喹啉(5.18g,17.5mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ7.36-7.40(m,1H),7.41-7.42(m,2H),7.46-7.55(m,2H),7.67-7.74(m,2H),7.77(d,J=6.0Hz,1H),7.83(d,J=8.8Hz,1H),7.94(d,J=8.4Hz,1H),8.00-8.04(m,1H),8.11(dd,J=1.4Hz及5.6Hz,1H)8.72(d,J=5.6Hz,1H)
MALDI-TOF MS:m/z 296([M+H]+)。
前驅體(2)的合成
依照下式,使C-N配位基(2)與氯化銥進行反應從而得到前驅體(2)。在氮氣氣氛下,在油浴中加熱1-(二苯並[b,d]呋喃-4-基)異喹啉(3.07g,10.4mmol)與2-乙氧基乙醇(180mL)的混合物。若溶液的溫度達到100℃,則加入氯化銥三水合物(1.21g,4.04mmol)與水(60mL)的混合物,在120℃下攪拌所得的反應混合物10小時。冷卻後,將水(150mL)添加到反應混合物,進行過濾從而回收沉澱物,並以適量的甲醇進行清洗,由此以69%的產率得到前驅體(2)(2.28g,1.40mmol)。因前驅體(2)為難溶性的固體,故不進行進一步的精製,而將其用於下述銥配合物的合成。
[化16]
C-N配位基(3)的合成
依照下式,合成2-(二苯並[b,d]噻吩-4-基)喹啉作為C-N配位基(3)。
[化17]
將二苯並[b,d]噻吩-4-基硼酸(1.60g,7.01mmol)、2-氯喹啉(1.27g,7.76mmol)、四(三苯基膦)鈀(0.665g,0.575mmol)及碳酸鉀(14.8g,107mmol)的混合物添加到1,2-二甲氧基乙烷(75mL)、乙醇(75mL)及水(75mL)的混合溶劑中,在氮氣氣氛下,在設定為100℃的油浴中,使其加熱回流18小時。冷卻後,以蒸發器餾去有機溶劑,並加入100mL的氯仿。以水和飽和食鹽水清洗該混合物,並加入適量硫酸鎂從而使其乾燥。通過過濾除去硫酸鎂後,以蒸發器餾去濾液的溶劑。以矽膠柱層析法(展開溶劑;氯仿:己烷=2.5:1(v/v))對得到的殘渣進行精製,從而以89%的產率得到2-(二苯並[b,d]噻吩-4-基)喹啉(1.94g,6.23mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ7.46-7.53(m,2H),7.58(dt,J=1.4及7.6Hz,1H),7.63(t,J=7.8Hz,1H),7.80(dt,J=1.4及7.6Hz,1H),7.86(d,J=7.8Hz,1H),7.99(m,1H),8.10(d,J=8.7Hz,1H),8.17(dd,J=1.4及7.6Hz,1H),8.21-8.24(m,1H),8.26(d,J=8.7Hz,1H),8.30(dd,J=0.9及7.8Hz,1H),8.42(d,J=7.8Hz,1H)。
MALDI-TOF MS:m/z 311(M+)。
前驅體(3)的合成
依照下式,使C-N配位基(3)與氯化銥進行反應從而得到前驅體(3)。
[化18]
在氮氣氣氛下,在油浴中加熱2-(二苯並[b,d]噻吩-4-基)喹啉(1.74g,5.58mmol)與2-乙氧基乙醇(100mL)的混合物。若溶液的溫度達到100℃,則加入氯化銥三水合物(1.24g,3.52mmol)與水(35mL)的混合物,在120℃下攪拌所得的反應混合物10小時。冷卻後,將水(90mL)添加到反應混合物,進行過濾從而回收沉澱物,並以適量的甲醇進行清洗,由此以89%的產率得到前驅體(3)(2.11g,1.24mmol)。該化合物為難溶性的固體。不進行進一步的精製,而用於下述銥配合物的合成。
C-N配位基(4)的合成
依照下式,合成1-(二苯並[b,d]噻吩-4-基)異喹啉作為C-N配位基(4)。
[化19]
將二苯並[b,d]噻吩-4-基硼酸(1.57g,6.88mmol)、1-氯異喹啉(1.24g,7.58mmol)、四(三苯基膦)鈀(0.688g,0.595mmol)及碳酸鉀(14.7g,106mmol)的混合物添加到1,2-二甲氧基乙烷(75mL)、乙醇(75mL)及水(75mL)的混合溶劑中,在氮氣氣氛下,在設定為100℃的油浴中,使其加熱回流18小時。冷卻後,以蒸發器餾去有機溶劑,並加入100mL的氯仿。以水和飽和食鹽水清洗該混合物,並加入適量硫酸鎂從而使其乾燥。通過過濾除去硫酸鎂後,以蒸發器餾去濾液的溶劑。以矽膠柱層析法(展開溶劑;氯仿:己烷=2:1(v/v))對得到的殘渣進行精製,從而以77%的產率得到1-(二苯並[b,d]噻吩-4-基)異喹啉(1.65g,5.30mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ7.44-7.53(m,3H),7.64(t,J=7.8Hz,1H),7.69-7.72(m,2H),7.74-7.79(m,2H),7.94(d,J=8.3Hz,1H),7.98(d,J=7.8Hz,1H),8.21-8.24(m,1H),8.29(dd,J=7.6Hz,1H),8.71(d,J=6.0Hz,1H)。
MALDI-TOF MS:m/z 311(M+)。
前驅體(4)的合成
依照下式,使C-N配位基(4)與氯化銥進行反應從而得到前驅體(4)。
[化20]
在氮氣氣氛下,在油浴中加熱1-(二苯並[b,d]噻吩-4-基)異喹啉(1.65g,5.30mmol)與2-乙氧基乙醇(100mL)的混合物。若溶液的溫度達到100℃,則加入氯化銥三水合物(1.06g,3.01mmol)與水(35mL)的混合物,在120℃下攪拌所得的反應混合物10小時。冷卻後,將水(90mL)添加到反應混合物中,進行過濾從而回收沉澱物,並以適量的甲醇進行清洗,由此以71%的產率得到前驅體(4)(1.60g,0.943mmol)。該化合物為難溶性的固體。不進行進一步的精製,而用於下述銥配合物的合成。
以下述方式使以上所合成的前驅體(1)至(4)與β-二酮化合物(A)、(B)進行反應,得到各銥配合物(1-A、1-B、1-X、2-A、2-B、2-X、3-A、3-B、3-X、4-A、4-B、4-X)。
銥配合物(1-A)的合成
依照下式,使前驅體(1)與β-二酮(A)進行反應從而得到銥配合物1-A。
[化21]
將前驅體(1)(0.482g,0.295mmol)、1,3-雙(3,5-二-叔丁基苯基)丙-1,3-二酮(0.223g,0.497mmol)及碳酸鈉(0.382g,3.60mmol)添加到2-乙氧基乙醇(100mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:3(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以17%的產率得到銥配合物1-A(106mg,0.0862mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.17(s,36H),5.75(s,1H),6.67(d,J=8.2Hz,2H),7.06(d,J=1.8Hz,4H),7.19-7.23(m,4H),7.28(d,J=8.2Hz,2H),7.31(t,J=1.8Hz,2H),7.37-7.42(m,4H),7.69(d,J=8.2Hz,2H),7.77-7.81(m,4H),8.37(d,J=9.2Hz,2H),8.52(d,J=9.2Hz,2H),9.31(d,J=9.2Hz,2H)
MALDI-TOF MS:m/z 1229([M+H]+)。
C73H67IrN2O4分析計算值:C,71.37;H,5.50;N,2.28。實際測量值:C,71.74;H,5.84;N,2.13。
銥配合物(1-B)的合成
依照下式,使前驅體(1)與β-二酮(B)進行反應從而得到銥配合物(1-B)。
[化22]
將前驅體(1)(0.981g,0.601mmol)、1,3-雙(5,5,8,8-四甲基-5,6,7,8-四氫萘-2-基)丙-1,3-二酮(0.537g,1.21mmol)及碳酸鈉(0.796g,7.51mmol)添加到2-乙氧基乙醇(200mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:1.6(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以29%的產率得到銥配合物1-B(426mg,0.348mmol)。
1H NMR(CDCl3):δ1.08(s,6H),1.10(s,6H),1.16(s,6H),1.18(s,6H),1.57(m,8H),5.79(s,1H),6.65(d,J=8.2Hz,2H),7.09(d,J=8.2Hz,2H),7.13-7.18(m,4H),7.21-7.29(m,6H),7.34-7.43(m,4H),7.69(d,J=8.2Hz,2H),7.77-7.79(m,4H),8.34(d,J=8.3Hz,2H),8.50(d,J=8.3Hz,2H),9.29(d,J=8.2Hz,2H).
MALDI-TOF MS:m/z 1225([M+H]+)。
C73H63IrN2O4分析計算值:C,71.60;H,5.19;N,2.29。實際測量值:C,71.72;H,5.52;N,2.15。
銥配合物(1-X)的合成
依照下式,使前驅體(1)與β-二酮(X)進行反應從而得到銥配合物1-X。
[化23]
將前驅體(1)(0.534g,0.327mmol)、1,3-二苯基丙-1,3-二酮(0.138g,0.615mmol)及碳酸鈉(0.394g,3.72mmol)添加到2-乙氧基乙醇(100mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合物,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:3)對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,從而以1.6%的產率得到銥配合物1-X(10.0mg,0.00996mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ5.94(s,1H),6.64(d,J=8.2Hz,2H),7.12-7.23(m,8H),7.28-7.30(m,4H),7.35(t,J=7.8Hz,2H),7.41(t,J=7.8Hz,2H),7.48(d,J=7.3Hz,4H),7.70(d,J=8.2Hz,2H),7.77-7.80(m,4H),8.32(d,J=8.2Hz,2H),8.50(d,J=8.7Hz,2H),9.26(d,J=8.7Hz,2H).
MALDI-TOF MS:m/z 1004(M+)。
銥配合物(2-A)的合成
依照下式,使前驅體(2)與β-二酮(A)進行反應從而得到銥配合物2-A。
[化24]
將前驅體(2)(0.484g,0.296mmol)、1,3-雙(3,5-二-叔丁基苯基)丙-1,3-二酮(0.249g,0.555mmol)及碳酸鈉(0.396g,3.74mmol)添加到2-乙氧基乙醇(100mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合物,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:2)對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以9.0%的產率得到銥配合物2-A(61.4mg,0.0500mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ1.08(s,36H),6.42(s,1H),6.60(d,J=8.7Hz,2H),7.22-7.26(m,2H),7.30-7.34(m,10H),7.36(t,J=1.8Hz,2H),7.49-7.52(m,2H),7.71-7.79(m,6H),7.90(d,J=8.7Hz,2H),8.61(d,J=6.4Hz,2H),9.18(d,J=8.7Hz,2H).
MALDI-TOF MS:1228(M+)。
C73H67IrN2O4分析計算值:C,71.37;H,5.50;N,2.28。實際測量值:C,71.21;H,5.72;N,2.04。
銥配合物(2-B)的合成
依照下式,使前驅體(2)與β-二酮(B)進行反應從而得到銥配合物(2-B)。
[化25]
將前驅體(1)(0.985g,0.603mmol)、1,3-雙(5,5,8,8-四甲基-5,6,7,8-四氫萘-2-基)丙-1,3-二酮(0.532g,1.20mmol)及碳酸鈉(0.791g,7.46mmol)添加到2-乙氧基乙醇(200mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:2(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以39%的產率得到銥配合物2-B(573mg,0.468mmol)。
1H NMR(CDCl3):δ1.08(s,6H),1.15(s,6H),1.16(s,6H),1.18(s,6H),1.57(m,8H),6.47(s,1H),6.49(d,J=1.8Hz,2H),7.13(d,J=8.2Hz,2H),7.32-7.36(m,4H),7.41(dd,J=1.8及8.2Hz,2H),7.49-7.54(m,6H),7.71-7.79(m,6H),7.90-7.93(m,4H),8.59(d,J=6.4Hz,2H),9.15(d,J=8.2Hz,2H).
MALDI-TOF MS:m/z 1224([M+H]+)。
C73H63IrN2O4分析計算值:C,71.60;H,5.19;N,2.29。實際測量值:C,71.72;H,5.45;N,2.11。
銥配合物(2-X)的合成
依照下式,使前驅體(2)與β-二酮(X)進行反應從而得到銥配合物2-X。
[化26]
將前驅體(2)(0.979g,0.600mmol)、1,3-二苯基丙-1,3-二酮(0.270g,1.20mmol)及碳酸鈉(0.788g,7.43mmol)添加到2-乙氧基乙醇(200mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合物,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿)對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,從而以5.7%的產率得到銥配合物1-X(68.7mg,0.0684mmol)。以上述方法合成的化合物的特性如下所示。
1H NMR(CDCl3):δ6.44(d,J=8.2Hz,2H),6.57(s,1H),7.18-7.24(m,4H),7.30-7.38(m,8H),7.50(d,J=8.2Hz,2H),7.53(d,J=6.4Hz,2H),7.69(dd,J=1.3及8.2Hz,4H),7.71-7.79(m,6H),7.91(d,J=8.2Hz,2H),8.58(d,J=6.4Hz,2H),9.15(d,J=8.2Hz,2H).
MALDI-TOF MS:1004(M+)
銥配合物(3-A)的合成
依照下式,使前驅體(3)與β-二酮(A)進行反應從而得到銥配合物(3-A)。
[化27]
將前驅體(3)(1.02g,0.601mmol)、1,3-雙(3,5-二-叔丁基苯基)丙-1,3-二酮(0.536g,1.19mmol)及碳酸鈉(0.790g,7.45mmol)添加到2-乙氧基乙醇(200mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1.6:1(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以32%的產率得到銥配合物3-A(488mg,0.387mmol)。
1H NMR(CDCl3):δ1.18(s,36H),5.68(s,1H),6.87(d,J=8.2Hz,2H),7.01(d,J=1.8Hz,4H),7.19(dt,J=1.4及8.2Hz,2H),7.31(t,J=1.8Hz,2H),7.35-7.41(m,6H),7.46(d,J=8.2Hz,2H),7.81(d,J=6.8Hz,2H),7.92(dd,J=1.4及8.2Hz,2H),7.97(dd,J=1.4及6.9Hz,2H),8.43(d,J=8.7Hz,2H),8.46(d,J=8.7Hz,2H),8.83(d,J=8.7Hz,2H).
MALDI-TOF MS:m/z 1260(M+)。
C73H67IrN2O2S2分析計算值:C,69.55;H,5.36;N,2.22。實際測量值:C,69.55;H,5.36;N,2.54。
銥配合物(3-B)的合成
依照下式,使前驅體(3)與β-二酮(B)進行反應從而得到銥配合物(3-B)。
[化28]
將前驅體(3)(1.02g,0.601mmol)、1,3-雙(5,5,8,8-四甲基-5,6,7,8-四氫萘-2-基)丙-1,3-二酮(0.532g,1.20mmol)及碳酸鈉(0.787g,7.43mmol)添加到2-乙氧基乙醇(200mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:1.6(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以37%的產率得到銥配合物3-B(558mg,0.444mmol)。
1H NMR(CDCl3):δ1.08(s,6H),1.10(s,6H),1.16(s,6H),1.18(s,6H),1.57(m,8H),5.80(s,1H),6.65(d,J=8.2Hz,2H),7.09(d,J=8.2Hz,2H),7.13-7.16(m,4H),7.18-7.30(m,6H),7.34-7.42(m,4H),7.69(d,J=8.2Hz,2H),7.77-7.90(m,4H),8.34(d,J=8.7Hz,2H),8.50(d,J=8.7Hz,2H),9.29(d,J=8.7Hz,2H).
MALDI-TOF MS:m/z 1256(M+)。
C73H63IrN2O2S2分析計算值:C,69.77;H,5.05;N,2.23。實際測量值:C,69.49;H,5.25;N,2.54。
銥配合物(3-X)的合成
依照下式,使前驅體(3)與β-二酮(X)進行反應從而得到銥配合物(3-X)。
[化29]
將前驅體(3)(0.768g,0.453mmol)、1,3-二苯基丙-1,3-二酮(0.271g,1.21mmol)及碳酸鈉(0.592g,5.59mmol)添加到2-乙氧基乙醇(150mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=2:1(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,從而以5.7%的產率得到銥配合物3-X(53.8mg,0.0519mmol)。
1H NMR(CDCl3):δ5.87(s,1H),6.85(d,J=8.2Hz,2H),7.10-7.20(m,6H),7.32-7.51(m,14H),7.78(dd,1.8及8.2Hz,2H),7.92(dd,J=1.4及6.9Hz,2H),7.99(dd,J=1.4及6.9Hz,2H),8.36(d,J=8.7Hz,2H),8.44(d,J=8.7Hz,2H),8.74(d,J=8.7Hz,2H).
MALDI-TOF MS:m/z 1036(M+)
C57H35IrN2O2S2分析計算值:C,66.07;H,3.40;N,2.70。實際測量值:C,66.20;H,3.51;N,2.61。
銥配合物(4-A)的合成
依照下式,使前驅體(4)與β-二酮(A)進行反應從而得到銥配合物(4-A)。
[化30]
將前驅體(4)(0.548g,0.323mmol)、1,3-雙(3,5-二-叔丁基苯基)丙-1,3-二酮(0.296g,0.660mmol)及碳酸鈉(0.454g,4.28mmol)添加到2-乙氧基乙醇(100mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:1(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以29%的產率得到銥配合物4-A(234mg,0.186mmol)。
1H NMR(CDCl3):δ1.07(s,36H),6.41(s,1H),6.59(d,J=8.2Hz,2H),7.22-7.26(m,2H),7.29(d,J=1.8Hz,4H),7.31-7.35(m,6H),7.49-7.52(m,4H),7.71-7.77(m,6H),7.90(d,J=8.2Hz,2H),8.61(d,J=5.9Hz,2H),9.18(d,J=8.2Hz,2H).
MALDI-TOF MS:m/z 1261([M+H]+).
C73H67IrN2O2S2分析計算值:C,69.55;H,5.36;N,2.22。實際測量值:C,69.38;H,5.33;N,2.37。
銥配合物(4-B)的合成
依照下式,使前驅體(4)與β-二酮(B)進行反應從而得到銥配合物(4-B)。
[化31]
將前驅體(4)(1.02g,0.601mmol)、1,3-雙(5,5,8,8-四甲基-5,6,7,8-四氫萘-2-基)丙-1,3-二酮(0.532g,1.20mmol)及碳酸鈉(0.789g,7.44mmol)添加到2-乙氧基乙醇(200mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:2(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,由此以26%的產率得到銥配合物4-B(392mg,0.311mmol)。
1H NMR(CDCl3):δ0.96(s,6H),1.07(s,6H),1.14(s,6H),1.17(s,6H),6.47(d,J=7.7Hz,2H),6.48(s,1H),7.13(d,J=8.2Hz,2H),7.22-7.26(m,2H),7.31-7.35(m,4H),7.40(dd,J=1.8及8.7Hz,2H),7.49(d,J=6.8Hz,4H),7.53(d,J=1.8Hz,2H),7.70-7.78(m,6H),7.90(d,J=7.7Hz,2H),8.59(d,J=6.8Hz,2H),9.14(d,J=8.7Hz,2H).
MALDI-TOF MS:m/z 1256(M+)
C73H63IrN2O2S2分析計算值:C,69.77;H,5.05;N,2.23。實際測量值:C,70.10;H,5.31;N,2.54。
銥配合物(4-X)的合成
依照下式,使前驅體(4)與β-二酮(X)進行反應從而得到銥配合物(4-X)。
[化32]
將前驅體(4)(0.511g,0.301mmol)、1,3-二苯基丙-1,3-二酮(0.139g,0.620mmol)及碳酸鈉(0.405g,3.82mmol)添加到2-乙氧基乙醇(100mL)中,在氮氣氣氛下,在85℃下攪拌該混合物2小時。冷卻後,在減壓下餾去溶劑,並向殘渣中加入氯仿。以水和飽和食鹽水清洗所得的混合溶液,並加入適量硫酸鈉從而使其乾燥。通過過濾除去硫酸鈉後,以蒸發器餾去濾液的溶劑。以氧化鋁柱層析法(展開溶劑;氯仿:己烷=1:1(v/v))對所得的殘渣進行精製,更進一步以氯仿-甲醇進行重結晶,從而以5.0%的產率得到銥配合物4-X(31.1mg,0.0300mmol)。
1H NMR(CDCl3):δ6.45(d,J=7.7Hz,2H),6.58(s,1H),7.21(t,J=7.6Hz,4H),7.23-7.27(m,2H),7.32-7.37(m,6H),7.50(d,J=8.2Hz,2H),7.53(d,J=6.4Hz,2H),7.70(d,J=7.6Hz,4H),7.72-7.80(m,6H),7.92(dd,J=1.3及8.2Hz,2H),8.59(d,J=6.4Hz,2H),9.16(d,J=7.7Hz,2H).
MALDI-TOF MS:m/z 1036(M+)
C57H35IrN2O2S2分析計算值:C,66.07;H,3.40;N,2.70。實際測量值:C,66.07;H,3.69;N,2.70。
對以上所得的各銥配合物進行發光光譜、PL量子產率及熱分解特性的評價。另外,使用各銥配合物製作有機EL元件,並對其特性進行評價。
[發光(PL)光譜及PL量子產率的評價]
對於上述所得的各銥配合物,測定其發光(PL)光譜及PL量子產率ΦPL。使用「堀場製作所社」制的Fluorolog-3分光光度計來進行PL光譜的測定。使用「浜松ホトニクス社」制的C9920-12量子產率測定裝置來進行PL量子產率的測定。對於這些PL光譜及PL量子產率的評價,在作為介質的有機溶劑(二氯甲烷(CH2Cl2))中、以及在高分子薄膜(聚甲基丙烯酸甲酯,PMMA)中的兩者中進行評價。另外,關於溶液試樣,封入氬氣作為脫氧溶液進行測定,關於高分子薄膜試樣,在氮氣氣氛下進行測定。關於高分子薄膜試樣,在PMMA中摻雜4重量%的各銥配合物從而進行測定。結果顯示於下表。
[表1]
由上述表1可知,相較於傳統例中經β-二酮(X)所取代的銥配合物(1-X、2-X、3-X、4-X),具有經β-二酮(A)或(B)所取代的苯基的銥配合物會有PL量子產率ΦPL較高的傾向。
[熱重量分析(TG)]
對於銥配合物的熱分解特性,通過熱重量分析(TG)進行評價。使用「リガク社」制的TG8120熱重量分析裝置作為TG測定裝置,在氮氣流下(200mL/min),以升溫速度10℃/min將2mg銥配合物從約50℃加熱至約450℃,觀察此時試樣的重量變化。測定結果顯示於圖1及表2。將表2的重量減少率作為相對於初期重量的重量減少率。
[表2]
由上述表格及圖2的結果可知,相較於傳統例中的經β-二酮(X)所取代的銥配合物(1-X、2-X、3-X、4-X),具有經β-二酮(A)或(B)所取代的苯基的銥配合物具有熱穩定性較高的傾向。
[有機EL元件的製作與特性評價]
使用銥配合物1-A、2-A、1-X及2-X,以下述順序製作圖2所示的有機元件(1),並進行特性評價。
(a)空穴注入層(5)的形成
對ITO-玻璃基板(「三容真空工業」制,ITO,膜厚150nm)實施圖案化處理,並進行清洗,由此製備陽極(2)。接著,以臭氧對ITO薄膜進行表面處理。表面處理後,通過旋轉塗布法迅速地使空穴注入材料成膜在ITO膜上,在120℃下燒制1小時,由此形成厚度為40nm的空穴注入層(5)。使用包含PEDOT與PSS的導電性聚合物(Heraeus Clevios制,P VP CH8000)作為空穴注入材料。
(b)發光層(4)的形成
使聚(9-乙烯基咔唑)(PVCz,Sigma-Aldrich制,數均分子量Mn為25000至50000,通過從THF-甲醇再沉澱進行精製)、2-(4-聯苯基)-5-(4-叔丁基苯基)-1,3,4-噁二唑(PBD)及銥配合物1-A溶解於脫水甲苯中,並以膜過濾器(メルクミリポア制,0.2μm Millex-FG)進行過濾,由此調製發光層用墨液Ink(1-A)。將PVCz、PBD及銥配合物的重量比設為10:3.0:1.0,相對於10mg的PVCz,使用0.7mL的甲苯作為墨液溶劑。使用所得的發光層用墨液Ink(1-A),通過旋轉塗布法,使其成膜於空穴注入層(5)上,並在120℃燒制1小時,由此形成厚度為80nm的發光層4。
(c)電子注入層(6)及陰極(3)的形成
使用陰影掩模,通過真空沉積形成作為電子注入材料的氟化銫薄膜(電子注入層(6),厚度為1nm),接著,製作鋁的薄膜(陰極(3),厚度為250nm)。此時,以使發光部的面積為10mm2(2mm×5mm)的方式來製作電子注入層(6)及陰極(3)。如此地,完成有機EL元件EL(1-A)。
使用各銥配合物(1-B、1-X、2-A、2-B、2-X、3-A、3-B、3-X、4-A、4-B、4-X)代替銥配合物1-A,調製各發光層用墨液Ink。除了使用該發光層用墨液Ink以外,以與上述相同的順序,得到各有機EL元件EL。
使用紫外線硬化樹脂,將以上述步驟所得的有機EL元件密封於中空玻璃中,從而製作有機EL特性評價用的樣品。
通過亮度光分布特性測定裝置(「浜松ホトニクス社」制、C-9920-11)來測定EL光譜、最大亮度Lmax(cd/m2)、最大外部量子效率ηext,max(%)及CIE表色系統(x,y)等有機EL元件特性。
表3顯示出有機EL元件的EL光譜的峰值波長λEL(nm)、最大亮度Lmax(cd/m2)、最大外部量子效率ηext,max(%)、最大電流效率ηj,max(cd/A)、最大功率效率ηp,max(lm/W)及CIE表色系統(x,y)的結果。關於Lmax及ηext,max,在內也一併顯示測定時的施加電壓(V)。另外,發光開始電壓Vturn-on表示亮度達到1cd/m2的電壓。
另外,圖3為各有機EL元件的電致發光(EL)光譜。EL光譜在最大亮度Lmax中進行測定。
[表3]
由上述結果可知,對於使用配合物1-A、1-B、2-A、2-B、3-A、3-B、4-A、4-B所製作的有機EL元件,其成為可顯示出與使用配合物1-X、2-X、3-X、4-X所製作的EL同等以上的有機EL特性的元件。
[工業實用性]
本發明的有機銥配合物的量子效率高,適合作為有機EL元件的發光材料。另外,該配合物的耐熱性高,有助於延長有機EL元件的壽命。