一種阿加曲班中間體(2R,4R)-4-甲基哌啶-2-甲酸乙酯的合成方法與流程
2023-05-20 13:17:56 2
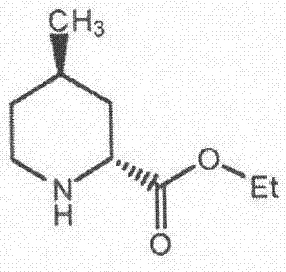
本發明屬於醫藥合成領域,具體涉及一種阿加曲班中間體(2r,4r)-4-甲基哌啶-2-甲酸乙酯的合成方法。
背景技術:
自1978年日本mitsubishi公司首次報導阿加曲班的抗凝血活性以來,眾多科研工作者對其化學合成、生物活性以及臨床應用進行了深入的研究。1990年首次在日本上市,2000年經美國fda批准上市,2002年在我國上市。阿加曲班可作為治療和預防血栓藥劑和血小板凝聚抑制劑、治療慢性動脈堵塞和治療腦血栓等藥物。阿加曲班的化學結構式如下:
在阿加曲班的結構中含有一個重要的反式哌啶酸結構單元:(2r,4r)-4-甲基哌啶-2-甲酸。在製備阿加曲班過程中作為(2r,4r)-4-甲基哌啶-2-甲酸的衍生物,(2r,4r)-4-甲基哌啶-2-甲酸乙酯是重要中間體,其化學結構式如下:
現有技術中有眾多製備(2r,4r)-4-甲基哌啶-2-甲酸乙酯的方法,例如us6440417中介紹了以手性銠催化劑作用下的不對稱氫化的方法來製備(2r,4r)-4-甲基哌啶-2-甲酸乙酯的方法,該方法使用了價格昂貴的手性銠催化劑,因而極大限制了規模化生產;文獻1agami,c.等,europeanjournaloforganicchemistry,2001,2385-2389中介紹了以手性苯甘氨醇衍生物為反應底物合成(2r,4r)-4-甲基哌啶-2-甲酸乙酯的方法,該方法起始原料不易得到,且使用了價格昂貴的鉑催化劑,因而不利於規模化生產;文獻2alegret,c.等,journaloforganicchemistry,2007,72,7688-7692中介紹了以手性環氧化物為反應底物合成(2r,4r)-4-甲基哌啶-2-甲酸乙酯方法,但該方法仍存在起始原料不易得,合成路線較長,且使用了價格昂貴的grubbs催化劑和氧化鉑催化劑等困難,導致該過程難以進行規模化生產。
jp53-73569、jp56-104866中介紹了以4-甲基哌啶為反應底物合成4-甲基哌啶-2-甲酸乙酯方法,該方法得到的產物為4-甲基哌啶-2-甲酸乙酯的多個異構體的混合物,需要藉助化學拆分劑進行化學拆分才能得到(2r,4r)-4-甲基哌啶-2-甲酸乙酯,收率較低,難以實現規模化生產;cn103641772a中公開了一種合成4-甲基哌啶-2-甲酸乙酯方法,具體反應路線為:但該方法兩布收率僅為50%左右,相對收率仍比較低,有較大的提高空間。發明人著重在cn103641772a的基礎上對該路線進行了改進,以提高反應的總收率,降低生成成本。
技術實現要素:
本發明旨在克服現有技術的不足並提供一種反應條件易控,成本低,尤其是收率高,操作過程簡便,適合於規模化生產的(2r,4r)-4-甲基哌啶-2-甲酸乙酯的合成方法。
為解決上述技術問題,本發明的技術方案如下:
一種(2r,4r)-4-甲基哌啶-2-甲酸乙酯化合物的製備方法,可按如下步驟依次實施:
(1)在銠催化劑和草酸鐵作用下,對(2r)-4-甲基-1-((s)-1-苯乙基)-1,2,3,6-四氫吡啶-2-甲酸乙酯進行催化加氫,得到(2r,4r)-4-甲基-1-((s)-1-苯乙基)-2-哌啶甲酸乙酯;
(2)在鈀催化劑和草酸鐵及氫氣作用下,脫去苄基,得到(2r,4r)-4-甲基哌啶-2-甲酸乙酯。
優選,本發明所述步驟(1)中,銠催化劑為銠與γ-氧化鋁、二氧化矽、輕質碳酸鈣、硫酸鋇、活性炭等中的一種或兩種以上混合物。
優選,本發明所述步驟(1)中,銠催化劑的用量為4-甲基(1-甲基苄基)四氫吡啶-2-甲酸乙酯重量的0.1%~10%,草酸鐵的用量(重量)約為銠催化劑的五分之一。
優選,本發明所述步驟(2)中,鈀催化劑為鈀-碳或氫氧化鈀-碳,更優選為優選為鈀-碳催化劑。
優選,本發明所述鈀催化劑用量為(2r,4r)-4-甲基-1-((s)-1-苯乙基)-2-哌啶甲酸乙酯重量的0.1%~20%,草酸鐵的用量(重量)約為鈀催化劑的五分之一。
優選,本發明所述步驟(1)-(2)中,反應壓力為0.1~10mpa,更優選為優選為0.1~5mpa。
優選,本發明所述反應溫度為20~100℃。
優選,本發明所述反應時間為0.5~50小時。
優選,本發明還包括過濾、減壓濃縮、洗滌、乾燥、過濾、減壓濃縮、蒸餾等後處理步驟。
本發明合成(2r,4r)-4-甲基哌啶-2-甲酸乙酯的方法。以(2r)-4-甲基-1-((s)-1-苯乙基)-1,2,3,6-四氫吡啶-2-甲酸乙酯為原料,採用催化加氫還原雙鍵、催化脫苄基保護基、精餾純化等工藝過程完成了其合成。合成路線選用較經濟的負載銠催化劑,避免了昂貴手性銠催化劑的使用;脫苄基用負載鈀催化劑和草酸鐵完成,最後通過精餾分離純化,製得(2r,4r)-4-甲基哌啶-2-甲酸乙酯。該工藝過程原料易得,產物純度高,並且整條路線收率高,適合於規模化生產。
本發明所述的(2r,4r)-4-甲基哌啶-2-甲酸乙酯的合成方法,其主要效果為:發明人意外的發現在兩步反應的催化劑中加入少量的草酸鐵後整體的收率有了明顯的提高(比cn103641772a未添加草酸鐵的技術方案收率高),屬於預料不到的技術效果,尤其是草酸鐵和銠-二氧化矽催化劑組合使用時,收率相對於草酸鐵與其他銠催化劑(比如銠-氧化鋁催化劑、銠-碳催化劑、銠-碳酸鋇催化劑等)的組合更高,並且產物的純度較高達到99%以上。此外發明人還嘗試添加丙二酸鐵、草酸銅等物質,均未起到提高收率的效果。
附圖說明
圖1:阿加曲班的化學結構式圖;
圖2:(2r,4r)-4-甲基哌啶-2-甲酸乙酯化學結構式圖;
圖3:4-甲基哌啶-2-甲酸乙酯合成方法反應路線圖。
具體實施方式
實施例1一、(2r,4r)-4-甲基-1-((s)-1-苯乙基)四氫吡啶-2-甲酸乙酯的製備
將(2r)-4-甲基-1-((s)-1-苯乙基)-1,2,3,6-四氫吡啶-2-甲酸乙酯(625g,2.3mol)和乙醇(2l)加至5l高壓釜中,加入銠-二氧化矽催化劑(5%銠負載量,35g)和7g草酸鐵,通入h2,於35℃、1mpa反應10h,過濾,回收催化劑。反應液減壓濃縮,加入乙酸乙酯(750ml),飽和食鹽水洗滌(250mlx2),經無水硫酸鈉乾燥後過濾,濾液減壓濃縮後得無色透明液體產物(618g)。
液相色譜測定:(2r,4r)-4-甲基-1-((s)-1-苯乙基)-2-哌啶甲酸乙酯含量為92.4%,計算收率為90.6%;
二、(2r,4r)-4-甲基哌啶-2-甲酸乙酯的製備
將上述步驟中得到的產物粗品(618g)和乙醇(2ml)加入5l高壓釜中,再加入乙酸(45g)、鈀-碳催化劑(10%鈀負載量,15g)、3g草酸鐵,通入h2,於30℃、0.5mpa反應4h,過濾,回收催化劑。濾液減壓濃縮後加入乙酸乙酯(1l),依次用飽和碳酸鈉溶液(250mlx2)、飽和食鹽水(250mlx2)洗滌,經無水硫酸鈉乾燥後抽濾,濾液減壓濃縮,得淡黃色液體產物。
將得到的產物粗品精餾純化,收集89~90℃/10mmhg餾分,得到(2r,4r)-4-甲基-2-哌啶甲酸乙酯345g,收率:91%。
旋光度:(c=5,etoh)
以上過程總收率:82.45%。
液相色譜測定:(2r,4r)-4-甲基-2-哌啶甲酸乙酯含量為99.3%。
實施例2
一、(2r,4r)-4-甲基-1-((s)-1-苯乙基)四氫吡啶-2-甲酸乙酯的製備
將(2r)-4-甲基-1-((s)-1-苯乙基)-1,2,3,6-四氫吡啶-2-甲酸乙酯(625g,2.3mol)和乙醇(2l)加至5l高壓釜中,加入銠-氧化鋁催化劑(5%銠負載量,35g)和7g草酸鐵,通入h2,於35℃、1mpa反應10h,過濾,回收催化劑。反應液減壓濃縮,加入乙酸乙酯(750ml),飽和食鹽水洗滌(250mlx2),經無水硫酸鈉乾燥後過濾,濾液減壓濃縮後得無色透明液體產物(610g)。
液相色譜測定:(2r,4r)-4-甲基-1-((s)-1-苯乙基)-2-哌啶甲酸乙酯含量為83.6%,計算收率為80.9%;
二、(2r,4r)-4-甲基哌啶-2-甲酸乙酯的製備
將上述步驟中得到的產物粗品(610g)和乙醇(2ml)加入5l高壓釜中,再加入乙酸(45g)、鈀-碳催化劑(10%鈀負載量,15g)、3g草酸鐵,通入h2,於30℃、0.5mpa反應4h,過濾,回收催化劑。濾液減壓濃縮後加入乙酸乙酯(1l),依次用飽和碳酸鈉溶液(250mlx2)、飽和食鹽水(250mlx2)洗滌,經無水硫酸鈉乾燥後抽濾,濾液減壓濃縮,得淡黃色液體產物。
將得到的產物粗品精餾純化,收集89~90℃/10mmhg餾分,得到(2r,4r)-4-甲基-2-哌啶甲酸乙酯302.5g,收率:89.3%。
旋光度:(c=5,etoh)
以上過程總收率:72.24%。
液相色譜測定:(2r,4r)-4-甲基-2-哌啶甲酸乙酯含量為98.5%。
實施例3
使用銠-碳催化劑(5%銠負載量)替代實施例1銠-二氧化矽催化劑,其他條件同實施例1,得步驟一收率為82.9%,步驟二收率為90.1%,液相色譜測定:(2r,4r)-4-甲基-2-哌啶甲酸乙酯含量為98.3%。
實施例4
使用銠-碳酸鋇催化劑(5%銠負載量)替代實施例1銠-二氧化矽催化劑,其他條件同實施例1,得步驟一收率為79.9%,步驟二收率為91.6%,液相色譜測定:(2r,4r)-4-甲基-2-哌啶甲酸乙酯含量為98.6%。