阿那曲波的惡唑烷酮中間體的晶型及其製備方法與流程
2023-04-25 18:47:57 4
本發明涉及阿那曲波,具體涉及一種阿那曲波的惡唑烷酮中間體的晶型及其製備方法。
背景技術:
阿那曲波是由美國默克公司研發的口服有效的小分子惡唑烷酮類選擇性CETP抑制劑,用於治療動脈粥樣硬化、冠心病等。鑑於本品與Torcetrapib具有相同的作用機制,研究者擔心Anacetrapib也會存在Torcetrapib樣的不良反應。為了探究CETP抑制劑作用靶點與不良反應的相關性,實驗人員開展了一系列研究。結果表明,Torcetrapib的不良反應與CETP抑制劑無關。以健康志願者和高血脂患者為對象的臨床研究表明,阿那曲波能安全、有效地調節冠心病以及冠心病高危患者的脂質水平,且對不良反應患者耐受。阿那曲波的惡唑烷酮中間體化學名稱為(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮。
US20143033380,WO2006014357A1,TW201436862A,US2006014413A1,WO2007136672,等報導了合成阿那曲波的惡唑烷酮中間體的製備方法,主要通過酮A作為中間體先還原後成環得到阿那曲波的惡唑烷酮中間體。
目前為止,未發現阿那曲波的惡唑烷酮中間體的晶型方面的報導。
技術實現要素:
針對現有技術的不足,本發明的目的是提供一種阿那曲波的惡唑烷酮中間體的晶型,本發明同時提供其簡單易行的製備方法。
本發明所述的阿那曲波的惡唑烷酮中間體的晶型,其結構式為:
該晶型的X射線粉末衍射圖的反射角2θ在4.36°(100)、8.48°(18.46)、11.28°(23.65)、14.87°(12.91)、15.65°(13.02)、17.02°(11.73)、18.78°(23.35)、20.19°(10.23)、22.70°(18.16)處具有特徵峰。
該晶型的X射線粉末衍射圖的反射角2θ在4.00°(4.01)、7.44°(4.27)、11.15°(8.05)、13.09°(2.35)、15.26°(3.26)、15.39°(2.98)、17.48°(4.65)、21.56°(2.61)、21.89°(5.85)、22.42°(4.09)、22.95°(4.66)、23.42°(3.40)、23.73°(2.58)、23.82°(4.09)、26.17°(2.46)、27.18°(2.15)處具有特徵峰。
上述兩組晶型數據中,第二組特徵峰是對第一組特徵峰的補充,上述括號內數據是譜線相對強度。
所述的阿那曲波的惡唑烷酮中間體的晶型的製備方法,是將阿那曲波的惡唑烷酮中間體粗品用溶劑溶解,然後用矽膠填料,用展開劑通過TLC分出阿那曲波的惡唑烷酮中間體溶液,濃縮後加入正庚烷,升溫至回流,直至全部溶解,過濾,冰浴冷卻至室溫,繼續攪拌析晶,再進行過濾,濾餅經乾燥至恆重,得到阿那曲波的惡唑烷酮中間體的晶型。
其中,
所述的溶劑為乙酸乙酯。
展開劑為正己烷、正庚烷、60~90石油醚、乙酸乙酯或二氯甲烷中的兩種的混合溶劑。
所述的矽膠為50~400目,優選為100~200目。
所述的冷卻方式為冰浴冷卻。
阿那曲波的惡唑烷酮中間體粗品的製備方法如下:
在氮氣保護下將A、異丙醇鋁、異丙醇和甲苯混合,升溫至50℃,在50℃下攪拌反應17h,降溫至25~30℃時加入氫氧化鉀,保溫反應,TLC檢測終點,反應完畢後,滴加鹽酸,調節pH=3~4,攪拌析晶,過濾,洗滌濾餅,得到阿那曲波的惡唑烷酮中間體的粗品;其中,A的結構式如下:
阿那曲波的惡唑烷酮中間體粗品製備的方程式為:
綜上所述,本發明的有益效果如下:
本發明是一種阿那曲波的惡唑烷酮中間體的新晶型;其製備方法簡單易行,易於實現。
附圖說明
圖1是實施例1中阿那曲波的惡唑烷酮中間體晶型的X-射線粉末衍射圖;
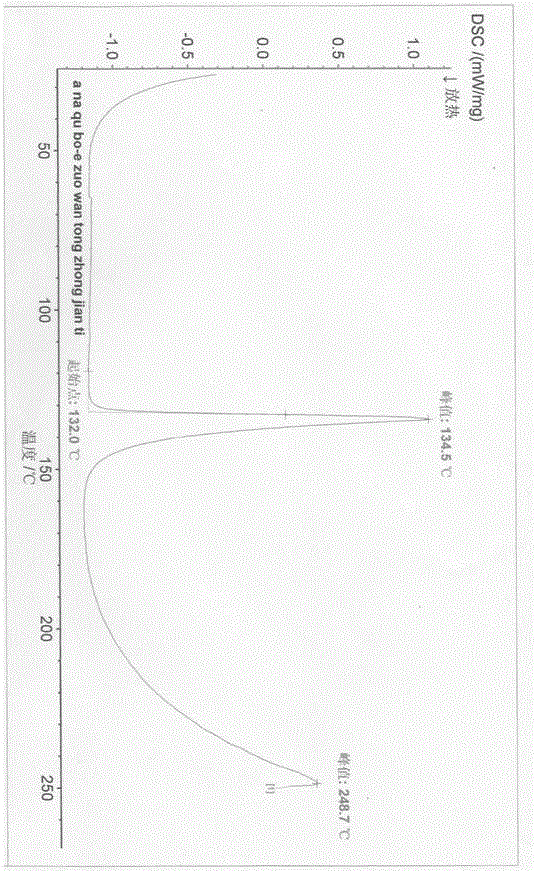
圖2是實施例1中阿那曲波的惡唑烷酮中間體晶型的差示掃描量熱圖;
圖3是實施例1中阿那曲波的惡唑烷酮中間體晶型的熱重分析圖。
具體實施方式
下面結合實施例對本發明做進一步說明。
實施例中涉及的A的結構式為:
實施例1
在250mL四口燒瓶中加入5.73g的A,然後加入32mL異丙醇和48mL甲苯,在室溫下氮氣置換0.5h。然後加入0.84g異丙醇鋁,同時升溫母液至50℃,然後在50℃攪拌反應過夜。水浴冷卻至25℃,然後向母液中分批加入1.4g氫氧化鉀,繼續在25℃保溫反應,TLC檢測終點。然後向母液中滴完3mL濃鹽酸,然後加入30mL水,在室溫下攪拌析晶1h。過濾,100mL純化水洗滌濾餅,在60℃,0.08MPa下真空乾燥2h,得到4.2g(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮粗品。
將上述所得粗品用10mL乙酸乙酯溶解。稱取50g 50~100目的矽膠,1000mL 1:1的正己烷和二氯甲烷作為展開劑。TLC選取目標產物溶液,濃縮得到3.65g粗品。然後粗品用50mL正庚烷加熱至80℃溶解,如果不能全溶,需補加正庚烷直至全部溶解。熱濾,濾餅在0℃析晶2h。抽濾,濾餅在60℃,0.08MPa下,真空乾燥2h至恆重,得到3.14g(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮。
本發明的晶型是化學純的,通過HPLC分析,歸一化純度≥99.9%,旋光-103.3,ee值為99.54%,外觀為白色片狀結晶。實施例1中,(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮晶型的X-射線粉末衍射圖(PXRD)如圖1所示,差示掃描量熱圖(DSC)如圖2所示,熱重分析圖(TGA)如圖3所示。
實施例2
在500mL四口燒瓶中加入16.81g的A,然後加入90mL異丙醇和110mL甲苯,在室溫下氮氣置換0.5h。然後加入2.45g異丙醇鋁,同時升溫母液至50℃,然後在50℃攪拌反應過夜。水浴冷卻至30℃,然後向母液中分批加入4.17g氫氧化鉀,繼續在30℃保溫反應,TLC檢測終點。然後向母液中滴完10mL濃鹽酸,然後加入90mL水,在室溫下攪拌析晶1h。過濾,100mL純化水洗滌濾餅,在60℃,0.08MPa下真空乾燥2h,得到15.54g(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮粗品。
將上述所得粗品用30mL乙酸乙酯溶解。稱取200g 100~200目的矽膠,3000mL 1:1的正庚烷和乙酸乙酯作為展開劑。TLC選取目標產物溶液,濃縮得到12.74g粗品。然後粗品用150mL正庚烷加入至80℃溶解,如果不能全溶,需補加正庚烷直至全部溶解。熱濾,濾餅在0℃析晶2h。抽濾,濾餅在60℃,0.08MPa下,真空乾燥2h至恆重,得到10.53g(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮。
實施例3
在500mL四口燒瓶中加入15.35g的A,然後加入80mL異丙醇和100mL甲苯,在室溫下氮氣置換0.5h。然後加入2.22g異丙醇鋁,同時升溫母液至50℃,然後在50℃攪拌反應過夜。水浴冷卻至28℃,然後向母液中分批加入3.96g氫氧化鉀,繼續在28℃保溫反應,TLC檢測終點。然後向母液中滴完9mL濃鹽酸,然後加入80mL水,在室溫下攪拌析晶1h。過濾,100mL純化水洗滌濾餅,在60℃,0.08MPa下真空乾燥2h,得到12.89g(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮粗品。
將上述所得粗品用25mL乙酸乙酯溶解。稱取180g 300~400目的矽膠,2500mL 1:1的60~90石油醚和乙酸乙酯作為展開劑。TLC選取目標產物溶液,濃縮得到11.45g粗品。然後粗品用100mL正庚烷加入至80℃溶解,如果不能全溶,需補加正庚烷直至全部溶解。熱濾,濾餅在0℃析晶2h。抽濾,濾餅在60℃,0.08MPa下,真空乾燥2h至恆重,得到9.88g(4S,5R)-5-(3,5-雙(三氟甲基)苯基)-4-甲基-1,3-唑烷-2-酮。