一種活性聚氨酯醫用膠黏劑及其製備方法與流程
2023-05-14 12:06:21 5
本發明屬於醫用材料技術領域,具體涉及一種活性聚氨酯醫用膠黏劑及其製備方法。
背景技術:
我國「十二五」科技發展規劃中已明確將生物醫學材料列為發展的重點。作為一種幾乎關係到每個人身體健康的生物醫學材料,醫用膠黏劑在替代傳統縫合技術用於傷口癒合方面顯示出極為廣闊的應用前景,醫用膠黏劑在國內外都得到了迅速的發展。但是,目前應用較廣的α-氰基丙烯酸酯類和纖維蛋白類醫用膠黏劑等仍然存在著諸如柔韌性差、會分解有毒物質或粘合強度不高等弊端。因此,製備具有良好柔韌性和粘結性能、具有良好生物相容且可生物降解的醫用膠黏劑是十分有意義的。
大量的研究者專注於開發各種各樣的新型高分子醫用粘合劑材料,其中,聚氨酯膠黏劑被認為是解決這些問題的最有前景的生物醫學材料之一,聚氨酯膠黏劑具備優異的抗剪切強度和抗衝擊特性,適用於各種結構性粘合領域,並且具備優異的柔韌性。聚氨酯膠黏劑因為其中含有極性很強、化學活潑性很高的異氰酸酯和氨基甲酸酯基團,所以它與許多含有活潑氫的材料,如:泡沫塑料、紙張、玻璃、橡膠、塑料等都有著優良的化學粘合力。聚氨酯與被粘合材料之間產生的氫鍵作用會使分子內聚力增強,從而使粘結更加牢固。此外,聚氨酯膠黏劑還具有良好的韌性、可調節性、粘合工藝簡便、極佳的耐低溫性能以及優良的穩定性等特性,但是其在人體組織環境下的粘結性能及可生物降解性能尚有待於改進。
綜上所述,傳統醫用膠黏劑一般具有易分解有毒物質、柔韌性差和粘合強度不高等問題,因此製備有良好柔韌性和粘結性能、具有良好生物相容且可生物降解的醫用膠黏劑是十分有意義的。
技術實現要素:
針對以上現有技術存在的缺點和不足之處,本發明的首要目的在於提供一種活性聚氨酯醫用膠黏劑的製備方法。
本發明的另一目的在於提供一種通過上述方法製備得到的活性聚氨酯醫用膠黏劑。
本發明目的通過以下技術方案實現:
一種活性聚氨酯醫用膠黏劑的製備方法,包括如下製備步驟:
(1)稱取一定量的蓖麻油,加入聚乙二醇催化劑,升溫至50~99℃,然後加入到恆溫至50~99℃的naoh水溶液中,保溫攪拌反應20~90min,得到蓖麻油脂肪酸和甘油的混合液,分離得到蓖麻油脂肪酸;
(2)將步驟(1)得到的蓖麻油脂肪酸升溫至90~120℃,加入環氧大豆油,同時在有機酸存在的條件下加入胺類催化劑,繼續保溫攪拌反應6~12h,反應結束後,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到50~80℃,在惰性氣體保護和攪拌條件下,滴加到恆溫50~80℃的二異氰酸酯溶液中反應1~4h,反應結束後,減壓蒸餾除去反應體系中殘留的雜質及微量的異氰酸酯單體,得到活性聚氨酯低聚物;
(4)向步驟(3)的活性聚氨酯低聚物中加入包括穩定劑或阻聚劑的助劑攪拌混合均勻,即得活性聚氨酯醫用膠黏劑。
優選地,步驟(1)中所述分離得到蓖麻油脂肪酸的過程為:向反應完全後的混合液中加入鹽酸至混合液呈酸性,攪拌,靜置分層後進行分液,從下層液體中回收甘油,上層液體用熱水洗滌至中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸。
優選地,步驟(1)中聚乙二醇催化劑的加入量為蓖麻油質量的0.16%~0.42%,naoh的加入量為蓖麻油質量的10.0%~24.0%;步驟(2)中環氧大豆油加入量為蓖麻油質量的40.0%~64.0%,胺類催化劑加入量為蓖麻油質量的0.50%~1.48%;步驟(3)中二異氰酸酯加入量為蓖麻油質量的24.0%~50.0%;步驟(4)中助劑的加入量為蓖麻油質量的0.50%~1.00%。
優選地,步驟(1)和步驟(2)中所述攪拌的轉速為200~300r/min。
優選地,步驟(3)中所述的惰性氣體為氮氣。
優選地,所述聚乙二醇催化劑是指分子量為200、300、400、600的聚乙二醇中的至少一種。
優選地,所述的有機酸為甲酸、乙酸、丙酸中的至少一種。
優選地,所述的胺類催化劑為乙二胺、三乙胺、三乙醇胺中的至少一種。
優選地,所述的二異氰酸酯為二苯基甲烷二異氰酸酯、六亞甲基二異氰酸酯、甲苯二異氰酸酯、異佛爾酮二異氰酸酯中的至少一種。
步驟(4)中所述穩定劑或阻聚劑優選為氫醌、二氧化硫、三氟乙酸和對苯二酚中的至少一種。
一種活性聚氨酯醫用膠黏劑,通過上述方法製備得到。
本發明的原理為:
①以精製蓖麻油(co)為原料,以naoh為皂化劑進行皂化反應,製得具有均一組成和結構、含羥基及18個碳原子的蓖麻油脂肪酸(cofa)。其反應路線圖如圖1所示。
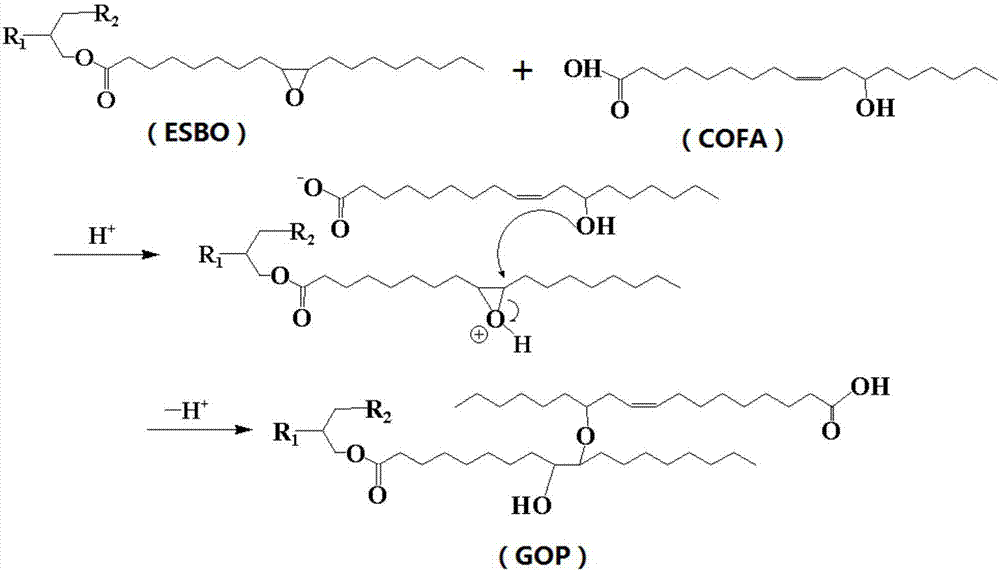
②在上述①合成的蓖麻油脂肪酸(cofa)的基礎上,加入環氧大豆油(esbo),加入胺類催化劑,在有機酸存在的條件下進行大豆油的環氧基團與蓖麻油的羥基的開環加成聚合反應。用1hnmr和ftir監控反應過程中環氧基團的反應情況,研究環氧基團與羥基的摩爾反應比對合成過程及產物性能的影響,並適當調整合成工藝,製得分子量及結構可控的綠色的植物油多元醇(gop)。其反應路線圖如圖2所示。
③在上述②合成的植物油多元醇(gop)的基礎上,加入高活性的二異氰酸酯(hdi),利用②中加入的胺類催化劑的催化作用,催化異氰酸酯的-nco基與多元醇的-oh基之間的聚合反應,避免了有毒的有機錫類催化劑(如常用的二月桂酸二丁基錫)的使用。用正丁胺滴定法監控反應過程中-nco基團的含量,當其達到設定值時,停止反應,然後採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,最後製得完全無毒、不含溶劑、端基為-nco基且官能度在3~5之間、相對分子量在2000~6000之間的活性聚氨酯低聚物(aopu)。其結構示意圖如圖3所示:
本發明的製備方法及所得到的產物具有如下優點及有益效果:
(1)本發明以可再生的生物質資源蓖麻油為原料,通過環氧大豆油的環氧基團與蓖麻油的羥基的反應合成了結構及分子量可控的綠色植物油多元醇(gop),並在此基礎上進一步製備分子鏈末端為-nco基的活性聚氨酯醫用膠黏劑,具有很好的柔韌性、粘結強度、生物相容性和生物可降解性。
(2)本發明的製備方法不使用有毒的有機金屬類催化劑和有機溶劑,是一種活性聚氨酯醫用膠黏劑的綠色製備方法。
(3)本發明的活性聚氨酯醫用膠黏劑單組份液體,其在潮溼空氣中(相對溼度rh≥85%)固化後的膠膜拉伸強度為2~10mpa,斷裂伸長率為150%~250%,撕裂強度為10~15kn/m,剝離強度高,20~50℃下固化;可抹塗或噴霧使用,在人體環境下固化時間小於30s,組織粘結強度大於11n/cm2,具有良好的生物相容性,不易引起炎症、異物反應、組織壞死或廣泛的纖維變性,使用後體表膠在7~10天隨皮膚角化脫落,體內應用膠在2~3個月內被逐漸降解吸收排出體外。
附圖說明
圖1為本發明蓖麻油脂肪酸的合成反應路線圖;
圖2為本發明植物油多元醇的合成反應路線圖;
圖3為本發明所得活性聚氨酯低聚物的結構示意圖。
具體實施方式
下面結合實施例對本發明作進一步詳細的描述,但本發明的實施方式不限於此。
實施例1
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至80℃,然後迅速加入到47.7g恆溫至80℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應36min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油;上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將39.1g得到的蓖麻油脂肪酸升溫至110℃,加入21.3g環氧大豆油,同時在乙酸存在的條件下加入0.45g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加恆溫到60℃的17.6g六亞甲基二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.32g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為65%,數均分子量為5300;固化後的膠膜拉伸強度為5.1mpa,斷裂伸長率為185%,撕裂強度為12.4kn/m。
實施例2
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至80℃,然後迅速加入到53.7g恆溫至80℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應20min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油;上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將38.8g得到的蓖麻油脂肪酸升溫至110℃,加入21.1g環氧大豆油,同時在乙酸存在的條件下加入0.45g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加到恆溫60℃的17.5g六亞甲基二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.31g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為65%,數均分子量為5200;固化後的膠膜拉伸強度為5.0mpa,斷裂伸長率為181%,撕裂強度為12.0kn/m。
實施例3
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至60℃,然後迅速加入到47.7g恆溫至60℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應80min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油;上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將39.0g得到的蓖麻油脂肪酸升溫至110℃,加入21.2g環氧大豆油,同時在乙酸存在的條件下加入0.45g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加到恆溫60℃的17.6g六亞甲基二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.32g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為65%,數均分子量為5000;固化後的膠膜拉伸強度為5.0mpa,斷裂伸長率為182%,撕裂強度為11.9kn/m。
實施例4
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至80℃,然後迅速加入到47.7g恆溫至80℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應36min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油;上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將39.1g得到的蓖麻油脂肪酸升溫至90℃,加入21.3g環氧大豆油,同時在乙酸存在的條件下加入0.45g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加到恆溫60℃的17.6g異佛爾酮二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.32g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為60%,數均分子量為3000;固化後的膠膜拉伸強度為4.2mpa,斷裂伸長率為161%,撕裂強度為9.9kn/m。
實施例5
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至80℃,然後迅速加入到47.7g恆溫至80℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應36min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油。上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將39.1g得到的蓖麻油脂肪酸升溫至110℃,加入21.3g環氧大豆油,同時在乙酸存在的條件下加入0.63g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加恆溫到60℃的17.6g六亞甲基二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.32g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為65%,數均分子量為5200;固化後的膠膜拉伸強度為5.2mpa,斷裂伸長率為186%,撕裂強度為12.5kn/m。
實施例6
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至80℃,然後迅速加入到47.7g恆溫至80℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應36min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油;上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將39.1g得到的蓖麻油脂肪酸升溫至110℃,加入18.0g環氧大豆油,同時在乙酸存在的條件下加入0.45g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加恆溫到60℃的17.6g六亞甲基二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.32g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為60%,數均分子量為4400;固化後的膠膜拉伸強度為4.6mpa,斷裂伸長率為170%,撕裂強度為10.2kn/m。
實施例7
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至80℃,然後迅速加入到47.7g恆溫至80℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應36min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油;上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將39.1g得到的蓖麻油脂肪酸升溫至110℃,加入21.3g環氧大豆油,同時在乙酸存在的條件下加入0.45g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加恆溫到60℃的22.0g六亞甲基二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.32g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為68%,數均分子量為4800;固化後的膠膜拉伸強度為5.0mpa,斷裂伸長率為160%,撕裂強度為11.0kn/m。
實施例8
本實施例的一種活性聚氨酯醫用膠黏劑的製備方法,具體步驟如下:
(1)稱取40.7g蓖麻油,加入0.12g聚乙二醇催化劑(其分子量為400),升溫至80℃,然後迅速加入到47.7g恆溫至80℃質量分數為14.6%的naoh水溶液中,保溫攪拌反應36min得到蓖麻油脂肪酸和甘油的混合物,向反應完全後的混合液中加入稀hcl溶液至混合液呈酸性,攪拌並倒入分液漏鬥中,靜置分層後進行分液,從下層液體中回收甘油;上層液體用熱水洗滌,至洗滌水呈中性,由於蓖麻油脂肪酸不溶於水,分液後即得蓖麻油脂肪酸;
(2)將39.1g得到的蓖麻油脂肪酸升溫至110℃,加入21.3g環氧大豆油,同時在乙酸存在的條件下加入0.45g三乙胺催化劑繼續保溫攪拌反應8h,反應結束後,停止加熱,冷卻至室溫,得到液態環氧大豆油多元醇;
(3)將步驟(2)的產物加熱到60℃,在通入氮氣和攪拌的條件下,滴加到恆溫60℃的17.6六亞甲基二異氰酸酯中,利用環氧大豆油多元醇合成過程中加入的三乙胺催化劑的作用進行反應2h,期間,根據粘度不同,隨時調控攪拌器轉速。反應結束後,採用減壓蒸餾技術,除去反應體系中殘留的雜質及微量的異氰酸酯單體,製得活性聚氨酯低聚物;
(4)向步驟(3)合成的活性聚氨酯低聚物中加入0.44g氫醌和對苯二酚混合攪拌均勻,即製得活性聚氨酯醫用膠黏劑。
本實施例所得活性聚氨酯醫用膠黏劑的固含量為65%,數均分子量為5000;固化後的膠膜拉伸強度為5.0mpa,斷裂伸長率為182%,撕裂強度為12.1kn/m。
上述實施例為本發明較佳的實施方式,但本發明的實施方式並不受上述實施例的限制,其它的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護範圍之內。