氯甲西泮重排產物的製備方法與流程
2023-09-14 08:49:05 8
本發明涉及一種氯甲西泮重排產物的製備方法,屬於藥物合成技術領域。
背景技術:
奧沙西泮、蘿拉西泮、氯甲西泮等苯並二氮雜卓類藥物已被廣泛用於治療失眠、抑鬱症和相關的疾病。其中,氯甲西泮最早於德國上市,且對其重排產物的質量研究對於原料藥生產和製劑生產具有更加重要深遠的意義。J.Org.Chem.,Vol.40,No.10,1508-1510,(1975)公開了一種氯甲西泮重排產物的製備方法,該方法以蘿拉西泮為主要原料,二氯亞碸、三甲氧基膦或三乙氧基膦、硫酸二甲酯、氫化鈉為其餘原料,總共進行了五步反應才得到氯甲西泮重排產物,反應中也出現了很多劇毒中間體,存在反應步驟過長、收率低、易造成環境汙染的問題,因此,有必要對其製備方法進行改進。
技術實現要素:
本發明的目的是提供一種氯甲西泮重排產物的製備方法,解決了當前製備方法中存在的製備步驟過長、使用劇毒試劑、汙染環境的問題,具有原料易得、操作簡單、反應安全性高、收率高的特點。
本發明所述的氯甲西泮重排產物的製備方法,包括以下步驟:
(1)先將氯甲西泮加入到有機溶劑中攪拌均勻,再加入氫氧化鈉固體進行反應,然後採用減壓蒸餾的方式分離出有機溶劑,得到殘留物;其中,反應方程式如下:
(2)向殘留物中加入純化水進行一次洗滌,過濾後再用純化水進行二次洗滌,真空乾燥後得到氯甲西泮重排產物;
其中,優選的技術方案如下:
步驟(1)中的有機溶劑為甲醇、乙醇、乙酸乙酯、乙腈中的任意一種,優選乙酸乙酯。
步驟(1)中氯甲西泮與氫氧化鈉固體的摩爾比為1:1.2~1.8。
步驟(1)中的反應溫度為20~60℃,反應時間為1~3h,優選2h。
步驟(2)中真空乾燥時的真空乾燥時的壓力為-0.08MPa,溫度為80℃,乾燥時間為2h。
本發明的有益效果如下:
本發明採用氯甲西泮作為原料,僅用一步反應便得到了氯甲西泮重排產物,而目前的製備工藝用了很多劇毒或者不穩定的原料,且反應步驟太長,收率太低;本發明的製備方法的原料轉化率接近100%,收率>97%,產生的廢水較少,具有原料易得、後處理簡單、收率高、反應安全性高的特點。
附圖說明
圖1是採用本發明的製備方法得到的氯甲西泮重排產物的氫譜圖。
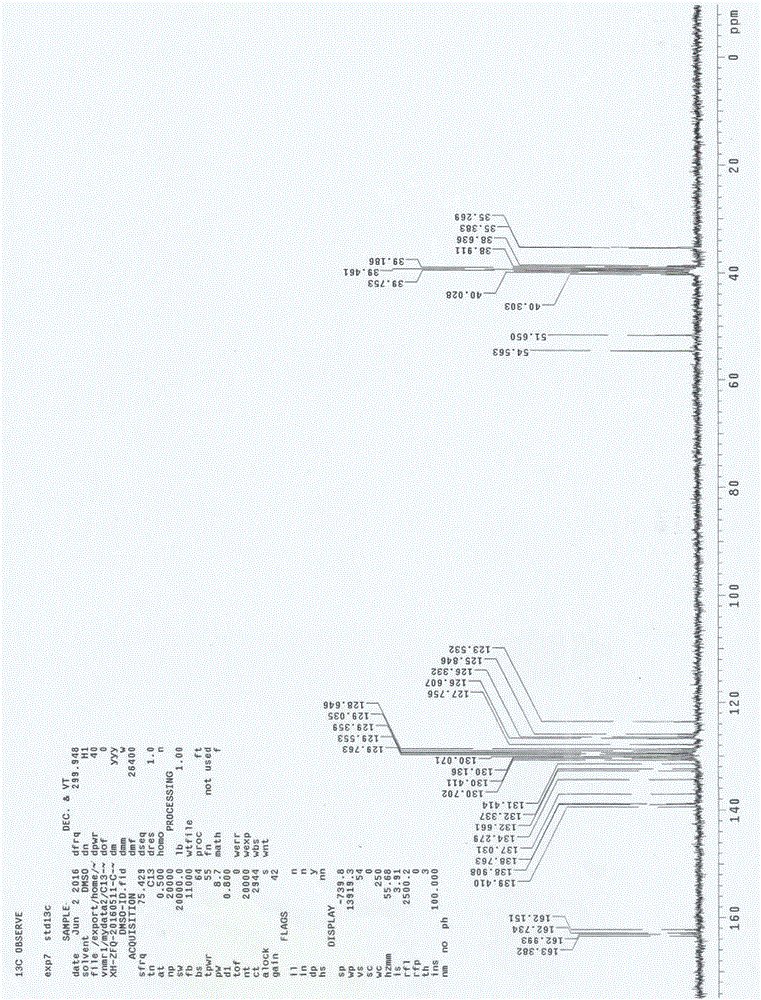
圖2是採用本發明的製備方法得到的氯甲西泮重排產物的碳譜圖。
圖3是採用本發明的製備方法得到的氯甲西泮重排產物的質譜圖。
具體實施方式
以下結合實施例對本發明做進一步描述。
實施例1
氯甲西泮重排產物的製備方法如下:
將0.1mol氯甲西泮加入到200mL無水乙醇中,室溫下攪拌均勻,然後加入0.12mol固體氫氧化鈉,在20℃下反應3h,減壓蒸餾出全部乙醇,得到殘留物,向殘留物中加入200mL純化水進行一次洗滌,過濾後再用100mL純化水進行二次洗滌,得到的固體於-0.08MPa、80℃下真空乾燥2h,得到32.39g氯甲西泮重排產物。經計算,產品收率為97.21%,純度為99.78%。
實施例2
氯甲西泮重排產物的製備方法如下:
將0.15mol氯甲西泮加到300mL乙酸乙酯中,室溫下攪拌均勻,然後加入0.225mol固體氫氧化鈉,在40℃下反應3h,減壓蒸餾出全部乙酸乙酯,得到殘留物,向殘留物中加入200mL純化水進行一次洗滌,過濾後再用100mL純化水進行二次洗滌,得到的固體於-0.08MPa、80℃下真空乾燥2h,得到48.73g氯甲西泮重排產物。經計算,本實施例的產品收率為97.5%,純度為99.85%。
實施例3
將0.2mol氯甲西泮加入到400mL乙腈中,室溫下攪拌均勻,然後加入0.36mol固體氫氧化鈉,在60℃下反應1h,減壓蒸餾出全部乙腈,得到殘留物,向殘留物中加入200mL純化水進行一次洗滌,過濾後再用100mL純化水進行二次洗滌,得到的固體於-0.08MPa、80℃下真空乾燥2h,得到64.91g氯甲西泮重排產物。經計算,產品收率為97.4%,純度為99.67%。