一種菌及其構建方法和應用與流程
2023-10-24 23:45:18 2
本發明屬於生物工程領域,涉及一種菌及其構建方法和應用。
背景技術:
石油基聚合物為人們的生產生活帶來了極大的便利。但是,隨著石化資源的日益減少,以及使用石化資源帶來的環境問題,尋找新的環境友好的非石油基聚合物受到越來越多的重視。生物基材料具有易生物降解、原料來源廣和易化學改良等優點,使其在很多領域中都有重要的應用。目前,聚對苯二甲酸丙二醇酯(PTT)和聚乳酸(PLA)是受關注度較高的兩種生物基材料。PTT是一種性能優異的聚酯類新型纖維,由對苯二甲酸和1,3-丙二醇(1,3-PD)縮聚而成,它綜合了聚對苯二甲酸乙二醇酯(PET)和聚對苯二甲酸丁二醇酯(PBT)的優勢,具有很好的柔軟性、蓬鬆性、抗汙性、彈性和可以常溫染色等特點,可以廣泛用於服裝、裝飾和工程塑料等各個領域。PLA也稱為聚丙交酯,是以乳酸(LAC)為主要原料聚合得到的聚合物,它具有較好的熱穩定性、抗溶劑性和生物相容性,可進行多種方式的加工,可用於製造包裝材料、纖維和非織造物等在服裝、建築、農業和醫療衛生等領域用途廣泛。此外,PLA製品具有良好的生物可降解性,使用後能被自然界中微生物完全降解,是公認的環境友好材料。
生物基材料PTT和PLA巨大的市場需求,推動了生物法生產生物基材料單體1,3-PD和光學純LAC的發展。目前生物法生產1,3-PD的方法主要分為兩類:第一類是利用基因工程大腸桿菌,以葡萄糖等糖類為底物,生產1,3-PD(例如CN201110093628;ZL200710104008);第二類是利用腸道細菌,以甘油為底物,生產1,3-PD(ZL200410100479;CN201180064621)。最近,由於生物柴油產業的發展,甘油作為其副產物,價格逐漸下降,是1,3-PD生產的理想底物。可利用甘油為底物生產1,3-PD的菌株主要包括肺炎克雷伯氏菌(Klebsiella peneumoniae)、產酸克雷伯氏菌(Klebsiella oxytoca)、弗氏檸檬酸桿菌(Citrobacterfreundii)和丁酸梭菌(Clostridium butyricum)等。用於生產PLA的LAC必須具有高光學純度,因此,研究者篩選得到了很多可生產光學純LAC的細菌,例如鼠李糖乳桿菌(Lactobacillus rhamnosus)、凝結芽孢桿菌(Bacillus coagulans)和土芽孢乳桿菌(Sporolactobacillus terrae)等野生細菌,都可以以糖類為原料生產高濃度的光學純L-乳酸(L-LAC)或D-乳酸(D-LAC)(ZL200710176057;ZL201210115365;CN201410022868)。
利用上述的菌株和生產方法,可實現1,3-PD和LAC中一種化合物的生物法生產,但是,目前尚無可同時生產1,3-PD和光學純LAC(L-LAC或D-LAC)的菌株。肺炎克雷伯氏菌和產酸克雷伯氏菌在以甘油為底物,生產1,3-PD的過程中,還會生產少量的2,3-丁二醇、乙醇、LAC、琥珀酸、乙酸和甲酸等副產物,雖然這兩種菌株發酵甘油的發酵液中,同時具有1,3-PD和LAC,但由於兩種化合物的濃度差距大(一般不在同一數量級)導致其中一種化合物只能被當做副產物處理,且醇類產物和酸類產物種類多,成分複雜,難以分離得到高純度的LAC,無法實現同時生產1,3-PD和LAC。在工業化大規模生產中,必須考慮產物濃度、轉化率,提取成本等因素,如果能夠利用一個菌株,以甘油為底物,以1,3-PD和光學純LAC(L-LAC或D-LAC)為主要產物,高轉化率的實現同時生產1,3-PD和光學純LAC(L-LAC或D-LAC),將大大降低生產成本,提高生產效率。
技術實現要素:
針對現有技術的不足,本發明提供一種菌,所述菌具有聯產1,3-丙二醇和L-乳酸的特性。聯產是指同時生產。
進一步地,所述菌是由野生菌改造獲得的人工菌。
進一步地,所述野生菌具有如下代謝途徑:
1)甘油→1,3-丙二醇;
所述野生菌還具有如下代謝途徑中一個或多個:
2)丙酮酸→α-乙醯乳酸,α-乙醯乳酸合成酶(budB)是催化該代謝途徑的酶;
3)α-乙醯乳酸→乙偶姻,α-乙醯乳酸脫羧酶(budA)是催化該代謝途徑的酶;
4)丙酮酸→乙酸,丙酮酸氧化酶(poxB)是催化該代謝途徑的酶;
5)乙醯輔酶A→乙醯磷酸,乙醯磷酸轉移酶(pta)是催化該代謝途徑的酶;
6)乙醯磷酸→乙酸,乙醯激酶(ackA)是催化該代謝途徑的酶;
7)乙醯輔酶A→乙醛,醛脫氫酶(adhE)是催化該代謝途徑的酶;
8)富馬酸→琥珀酸,富馬酸還原酶(frdA)是催化該代謝途徑的酶;
9)丙酮酸→D-乳酸,D-乳酸脫氫酶(dldh)是催化該代謝途徑的酶;
所述改造包括:阻斷所述代謝途徑中的2)-9)中的一個或多個。
進一步地,所述野生菌為產酸克雷伯氏菌(Klebsiella oxytoca)。
進一步地,所述野生菌為產酸克雷伯氏菌(Klebsiella oxytoca)PDL-0,所述產酸克雷伯氏菌(Klebsiella oxytoca)PDL-0於2016年4月8日保藏於中國典型培養物保藏中心,保藏登記號為CCTCC M 2016184。
進一步地,所述改造包括:通過抑制或去除酶的活性來阻斷所述代謝途徑中的2)-9)中的一個或多個。
進一步地,所述改造包括:通過改變酶的基因來抑制或去除酶的活性。
進一步地,所述改造包括:通過基因重組的辦法來改變酶的基因。
進一步地,編碼α-乙醯乳酸脫羧酶基因的序列如SEQ ID NO:1所示;
編碼α-乙醯乳酸合成酶基因的序列如SEQ ID NO:2所示;
編碼醛脫氫酶基因的序列如SEQ ID NO:3所示;
編碼乙酸激酶和乙醯磷酸轉移酶基因的序列如SEQ ID NO:4所示;
編碼丙酮酸氧化酶基因的序列如SEQ ID NO:5所示;
編碼富馬酸還原酶基因的序列如SEQ ID NO:6所示;
編碼D-乳酸脫氫酶基因序列如SEQ ID NO:7所示。
進一步地,所述人工菌的budA、budB、adhE、ackA-pta、poxB、frdA和dldh中的一種基因或者多種基因缺陷。
進一步地,所述基因缺陷通過同源重組的方法產生。
進一步地,所述改造還包括外源引入L-乳酸合成途徑。
進一步地,所述外源引入L-乳酸合成途徑指:將凝結芽孢桿菌(Bacillus coagulans)2-6的L-乳酸脫氫酶基因lldh通過質粒轉化到所述野生菌中,得到產L-乳酸的菌。
進一步地、如權利要求12所述的菌,其特徵在於,所述外源引入L-乳酸合成途徑指:將凝結芽孢桿菌(Bacillus coagulans)2-6的L-乳酸脫氫酶基因lldh替換所述野生菌中的budB基因,並通過質粒轉化到所述野生菌中,得到產L-乳酸的菌。
進一步地,所述人工菌的基因型包括ΔbudA::lldh ΔbudB ΔadhE ΔackA-ptaΔpoxB ΔfrdA Δdldh。
進一步地,所述人工菌的基因型是ΔbudA::lldh ΔbudB ΔadhE ΔackA-ptaΔpoxB ΔfrdA Δdldh。
從另一角度描述,所述人工菌是由所述野生菌的budA、budB、adhE、ackA-pta、poxB、frdA和dldh基因缺陷,並外源引入了lldh基因獲得;所述人工菌產1,3-PD和L-LAC,且1,3-PD和L-LAC的總轉化率超過90%。總轉化率計算公式為:總轉化率=1,3-丙二醇的摩爾轉化率+L-乳酸的摩爾轉化率;其中,1,3-丙二醇的摩爾轉化率=(1,3-PD的最終濃度×最終發酵液體積×甘油的摩爾質量92)/(消耗的甘油質量×1,3-PD的摩爾質量76);L-乳酸的摩爾轉化率=(L-LAC的最終濃度×最終發酵液體積×甘油的摩爾質量92)/(消耗的甘油質量×L-LAC的摩爾質量90)。
進一步地,budA、budB、adhE、ackA-pta、poxB、frdA和dldh基因缺陷的獲得採用同源重組的方法,且各基因缺陷的獲得不存在固定的先後順序;所述同源重組的方法是指通過PCR擴增以上所述基因的上遊同源片段和下遊同源片段,並將所述上遊同源片段和所述下遊同源片段構建到自殺質粒中轉化至大腸桿菌(Escherichia coli)中,得到供體菌;將所述供體菌與相應的受體菌進行雙親本雜交,使所述上遊同源片段和所述下遊同源片段與所述受體菌的基因組發生同源重組,從而獲得以上所述基因缺陷的菌株,即所述人工菌。
進一步地,所述野生菌是產酸克雷伯氏菌(Klebsiella oxytoca)PDL-0,所述產酸克雷伯氏菌(Klebsiella oxytoca)PDL-0於2016年4月8日保藏於中國典型培養物保藏中心,保藏登記號為CCTCC M 2016184。
進一步地,所述budA的DNA序列如SEQ ID NO:1所示;和/或所述budB的DNA序列如SEQ ID NO:2所示;和/或所述adhE的DNA序列如SEQ ID NO:3所示;和/或所述ackA-pta的DNA序列如SEQ ID NO:4所示;和/或所述poxB的DNA序列如SEQ ID NO:5所示;和/或所述frdA的DNA序列如SEQ ID NO:6所示;和/或所述dldh的DNA序列如SEQ ID NO:7所示;和/或所述lldh的DNA序列如SEQ ID NO:8所示。
在一較佳實施方式中,所述改造包括向所述野生菌中引入外源1,3-PD合成途徑和/或L-LAC合成途徑。
進一步地,所述引入外源1,3-PD合成途徑是將甘油脫水酶編碼基因dhaB及1,3-PD氧化還原酶編碼基因dhaT轉化到所述野生菌中。
進一步地,所述引入外源L-LAC合成途徑是將L-乳酸脫氫酶基因lldh通過質粒轉化到所述野生菌中,得到產L-乳酸的菌。
進一步地,所述外源引入的L-乳酸脫氫酶基因lldh來自於凝集芽孢桿菌(Bacillus coagulans)2-6。凝集芽孢桿菌(Bacillus coagulans)2-6為CN101173242A中的凝結芽孢桿菌(Bacillus coagulans)CASH,其保藏號為CGMCC No2184。
進一步地,所述改造還包括:通過基因重組的辦法來改變budA、budB、adhE、ackA-pta、poxB、frdA和/或dldh基因。
在一個較佳實施方式中,所述菌是產酸克雷伯氏菌(Klebsiella oxytoca)PLL,所述產酸克雷伯氏菌(Klebsiella oxytoca)PLL於2016年4月8日保藏於中國典型培養物保藏中心,保藏登記號為CCTCC M 2016186。
本發明提供的菌具有如下代謝途徑:
1)甘油→1,3-丙二醇;
2)甘油→丙酮酸→L-乳酸。
本發明提供的一種菌具有聯產1,3-丙二醇和L-乳酸的特性,其中聯產得到的:1,3-丙二醇的摩爾轉化率≥35%;L-乳酸的摩爾轉化率≥40%。
進一步地,聯產得到的:1,3-丙二醇的摩爾轉化率≥40%;和/或L-乳酸的摩爾轉化率≥50%。
本發明提供的一種菌具有聯產1,3-丙二醇和L-乳酸的特性,其中聯產得到的:1,3-丙二醇和L-乳酸的總轉化率超過90%。
本發明提供的一種菌具有聯產1,3-丙二醇和L-乳酸的特性,其中聯產得到的1,3-丙二醇和L-乳酸的質量比為1:0.1-10。進一步地,聯產得到的1,3-丙二醇和L-乳酸的質量比為1:0.2-5。進一步地,聯產得到的1,3-丙二醇和L-乳酸的質量比為1:0.5-2。
進一步地,本發明提供的所述菌來自於真菌中的麴黴屬、酵母屬、接合酵母屬、畢赤酵母屬、克魯維酵母屬、假絲酵母屬、漢遜酵母屬、德巴利酵母屬、毛黴屬、球擬酵母屬或鏈黴菌屬;或者來自細菌中的甲基菌屬、沙門氏菌屬、芽孢桿菌屬、假單胞菌屬、克雷伯氏菌、乳桿菌屬、腸桿菌屬、檸檬酸桿菌屬、暗桿菌屬、泥桿菌屬或者梭菌屬。
本發明還公開了一種如上所述的菌的構建方法,包括以下步驟:
步驟一、從土壤樣品中篩選能生產1,3-丙二醇和乳酸的產酸克雷伯氏菌;
步驟二、使得步驟一中得到的產酸克雷伯氏菌的α-乙醯乳酸脫羧酶基因、α-乙醯乳酸合成酶基因、醛脫氫酶基因、乙酸激酶和乙醯磷酸轉移酶基因、丙酮酸氧化酶基因、富馬酸還原酶基因和D-乳酸脫氫酶基因缺陷,得到過程菌株一;
步驟三、將凝結芽孢桿菌(Bacillus coagulans)2-6的L-乳酸脫氫酶基因插入到所述步驟二中得到的過程菌株一的α-乙醯乳酸脫羧酶基因在基因組中上遊的啟動子之後,得到具有L-乳酸脫氫酶活性的菌株,即所述菌。
進一步地,所述步驟一中得到的產酸克雷伯氏菌是產酸克雷伯氏菌(Klebsiella oxytoca)PDL-0,所述產酸克雷伯氏菌(Klebsiella oxytoca)PDL-0於2016年4月8日保藏於中國典型培養物保藏中心,保藏登記號為CCTCC M2016184。
進一步地,所述budA的DNA序列如SEQ ID NO:1所示;所述budB的DNA序列如SEQ ID NO:2所示;所述adhE的DNA序列如SEQ ID NO:3所示;所述ackA-pta的DNA序列如SEQ ID NO:4所示;所述poxB的DNA序列如SEQ ID NO:5所示;所述frdA的DNA序列如SEQ ID NO:6所示;所述dldh的DNA序列如SEQ ID NO:7所示;所述lldh的DNA序列如SEQ ID NO:8所示。
進一步地,所述步驟二包括以下步驟:
步驟2-1、使所述產酸克雷伯氏菌PDL-0 CCTCC M 2016184的α-乙醯乳酸脫羧酶基因和α-乙醯乳酸合成酶基因缺陷,得到α-乙醯乳酸脫羧酶和α-乙醯乳酸合成酶活性丟失的菌株,命名為產酸克雷伯氏菌PDL-1;
步驟2-2、使所述產酸克雷伯氏菌PDL-1的醛脫氫酶基因缺陷,得到醛脫氫酶活性丟失的菌株,命名為產酸克雷伯氏菌PDL-2;
步驟2-3、使所述產酸克雷伯氏菌PDL-2的乙酸激酶和乙醯磷酸轉移酶基因缺陷,得到乙酸激酶和乙醯磷酸轉移酶活性丟失的菌株,命名為產酸克雷伯氏菌PDL-3;
步驟2-4、使所述產酸克雷伯氏菌PDL-3的丙酮酸氧化酶基因缺陷,得到丙酮酸氧化酶活性丟失的菌株,命名為產酸克雷伯氏菌PDL-4;
步驟2-5、使所述產酸克雷伯氏菌PDL-4的富馬酸還原酶基因缺陷,得到富馬酸還原酶活性丟失的菌株,命名為產酸克雷伯氏菌PDL-5;
步驟2-6、使所述產酸克雷伯氏菌PDL-5的D-乳酸脫氫酶基因缺陷,得到D-乳酸脫氫酶基因活性丟失的菌株,命名為產酸克雷伯氏菌PDL-6;
經過以上步驟後得到了α-乙醯乳酸脫羧酶基因、α-乙醯乳酸合成酶基因、醛脫氫酶基因、乙酸激酶基因、乙醯磷酸轉移酶基因、丙酮酸氧化酶基因、富馬酸還原酶基因和D-乳酸脫氫酶基因缺陷的所述過程菌株一;
所述α-乙醯乳酸脫羧酶基因序列如SEQ ID NO:1所示;所述α-乙醯乳酸合成酶基因序列如SEQ ID NO:2所示;所述醛脫氫酶基因序列如SEQ ID NO:3所示;所述乙酸激酶和乙醯磷酸轉移酶基因序列如SEQ ID NO:4所示;所述丙酮酸氧化酶基因序列如SEQ ID NO:5所示;所述富馬酸還原酶基因序列如SEQ ID NO:6所示;所述D-乳酸脫氫酶基因序列如SEQ ID NO:7所示。
進一步地,所述步驟三中的凝結芽孢桿菌2-6的L-乳酸脫氫酶基因序列如SEQ ID NO:8所示。
進一步地,所述步驟二中,使得產酸克雷伯氏菌PDL-0 CCTCC M 2016184的α-乙醯乳酸脫羧酶基因、α-乙醯乳酸合成酶基因、醛脫氫酶基因、乙酸激酶和乙醯磷酸轉移酶基因、丙酮酸氧化酶基因、富馬酸還原酶基因和D-乳酸脫氫酶基因缺陷是通過PCR擴增以上所述基因的上遊同源片段和下遊同源片段,並將所述上遊同源片段和所述下遊同源片段構建到自殺質粒pKR6K上,並轉化至大腸桿菌(Escherichia coli)S17-1(λpir)中,得到供體菌;將所述供體菌與相應的受體菌進行雙親本雜交,使所述上遊同源片段和所述下遊同源片段與所述受體菌的基因組發生同源重組,從而獲得以上所述基因缺陷的菌株;所述受體菌包括所述產酸克雷伯氏菌PDL-0 CCTCC M 2016184、所述產酸克雷伯氏菌PDL-1、所述產酸克雷伯氏菌PDL-2、所述產酸克雷伯氏菌PDL-3、所述產酸克雷伯氏菌PDL-4和所述產酸克雷伯氏菌PDL-5;
所述步驟三中,將凝結芽孢桿菌(Bacillus coagulans)2-6的L-乳酸脫氫酶基因lldh插入到所述產酸克雷伯氏菌PDL-6的α-乙醯乳酸脫羧酶基因在基因組中上遊的啟動子之後,是通過PCR擴增所述α-乙醯乳酸脫羧酶基因在基因組中上遊同源片段和下遊同源片段,以及凝結芽孢桿菌2-6的L-乳酸脫氫酶基因片段後,構建到自殺質粒pKR6K上,並轉化至大腸桿菌S17-1(λpir)中,得到帶有pKR6K-ΔbudA::lldh的供體菌;將所述帶有pKR6K-ΔbudA::lldh的供體菌與所述產酸克雷伯氏菌PDL-6進行雙親本雜交,得到所述菌。
本發明還提供另一種如上所述的菌的構建方法,其特徵在於,通過基因工程改造向菌株中引入外源1,3-PD合成途徑和/或外源L-LAC合成途徑。
進一步地,所述引入外源1,3-PD合成途徑指:將甘油脫水酶編碼基因dhaB及1,3-PD氧化還原酶編碼基因dhaT通過質粒轉化到所述菌株中,得到菌株A,所述菌株A產1,3-PD。
進一步地,所述引入外源1,3-PD合成途徑指:選用產酸克雷伯氏菌中的1,3-PD合成途徑中的甘油脫水酶編碼基因dhaB及1,3-PD氧化還原酶編碼基因dhaT,進行PCR克隆後連接到質粒DNA pet-Duet上,並轉化到所述菌株中,得到菌株A,所述菌株A產1,3-PD。
進一步地,所述引入外源L-LAC合成途徑指:將L-乳酸脫氫酶基因lldh通過質粒轉化到所述菌株中,得到菌株B,所述菌株B產L-LAC。
進一步地,所述引入外源L-LAC合成途徑指:選用來自於凝結芽孢桿菌(Bacillus coagulans)2-6的L-乳酸脫氫酶基因lldh通過質粒轉化到所述菌株中,得到菌株B,所述菌株B產L-LAC。
進一步地,還包括如下操作:向引入外源1,3-PD合成途徑和/或引入外源L-LAC合成途徑的菌株,採用同源重組的方法產生budA、budB、adhE、ackA-pta、poxB、frdA和/或dldh基因缺陷的菌株。
本發明還涉及如上所述的菌在生產1,3-丙二醇或L-乳酸或聯產1,3-丙二醇和L-乳酸中的應用。
尤其是產酸克雷伯氏菌(Klebsiella oxytoca)PLL CCTCC M 2016186在生產1,3-丙二醇或L-乳酸或聯產1,3-丙二醇和L-乳酸中的應用。
本發明提供一種通過發酵如上所述的菌聯產1,3-丙二醇和L-乳酸的方法。通過發酵如上所述的菌聯產1,3-丙二醇和L-乳酸的方法,實際上是指如上所述的菌的應用。
進一步地,如上所述的聯產1,3-丙二醇和L-乳酸的方法,其特徵在於,所述菌是產酸克雷伯氏菌(Klebsiella oxytoca)PLL CCTCC M 2016186。
進一步地,發酵時通氣量為0-2.0vvm。進一步地,發酵時通氣量為1.0vvm。
進一步地,發酵時採用氫氧化鈣和水的混合乳液作為中和劑調節發酵液的pH。
進一步地,包括以下步驟:
步驟(1)、菌株選擇:選用產酸克雷伯氏菌(Klebsiella oxytoca)PLL CCTCC M 2016186;
步驟(2)、種子培養;
步驟(3)、發酵:發酵過程中,使用中和劑調節發酵液pH為5.5-7.5。
進一步地,包括以下步驟:
步驟(1)、菌株選擇:選用產酸克雷伯氏菌(Klebsiella oxytoca)PLL CCTCC M 2016186;
步驟(2)、種子培養:選用所述步驟(1)的菌株,在無菌條件下接種至甘油培養基中,培養溫度為25-40℃,搖床震蕩轉速為100-300rpm,培養時間為6-24小時,製得種子培養液;
步驟(3)、發酵:將所述步驟(2)中製得的種子培養液接種至裝有甘油培養基的發酵罐中,接種量v/v為0.5-10%,發酵溫度為25-40℃,通氣量為0.3-2.0vvm,攪拌轉速為50-400rpm,發酵過程中,使用中和劑調節發酵液pH為5.5-7.5;發酵方式為分批發酵或分批補料發酵,進行分批發酵時,當甘油培養基中的甘油耗盡時,停止發酵;進行分批補料發酵時,當甘油培養基中的甘油耗盡時,通過向發酵罐中補加400-800g/L的甘油溶液,控制發酵液中甘油濃度為5-40g/L,當發酵液中1,3-丙二醇或L-乳酸濃度不再升高時,停止發酵。
進一步地,所述步驟(2)中培養溫度為30-37℃,搖床震蕩轉速為150-250rpm,培養時間為10-16小時;所述步驟(3)中接種量v/v為2-6%,發酵溫度為30-37℃,通氣量為0.7-1.5vvm,攪拌轉速為150-300rpm,調節發酵液pH為6.0-7.0,發酵方式為分批補料發酵,補加的甘油溶液中甘油濃度為500-700g/L,控制發酵液中甘油濃度為10-30g/L;所述步驟(3)中的中和劑包括氫氧化鈉水溶液、氫氧化鉀水溶液、氨水水溶液,以及氫氧化鈣和水的混合乳液中的任一一種或者多種。
本發明的有益效果包括:
1、使用產酸克雷伯氏菌PLL CCTCC M 2016186,得到的發酵液中目標產物濃度高,副產物種類少且濃度低。可生產濃度超過70.7g/L的1,3-PD和濃度超過95.4g/L的L-LAC,1,3-PD和L-LAC的總轉化率超過90%,生產的L-LAC具有高光學純度(純度大於99.9%)。發酵液中僅能檢測到少量的副產物。
聯產1,3-PD和光學純LAC(L-LAC或D-LAC)的好處是:由於克雷伯氏菌代謝甘油屬於甘油歧化途徑,分為甘油氧化途徑和甘油還原途徑。甘油到1,3-丙二醇的轉化屬於甘油的還原途徑,轉化過程需要氧化途徑中產生的輔因子NADH和ATP來提供還原力及能量。乳酸在2004年被美國能源部評選為前三十種重點發展的平臺化合物之一,鑑於其廣闊的用途,在2010年又被選為最有前途的平臺化合物之一。乳酸是一種重要的單體,可以用來合成可生物降解、可生物相容且環境友好的生物多聚體-聚乳酸(PLA)。除了乳酸的經濟價值外,合成乳酸的過程中淨產1mol NADH,可以為合成1,3-PD提供還原力,從而達到還原力平衡的狀態;從甘油到乳酸的轉化過程中,是三碳到三碳的轉化,沒有碳損失,可以保證碳的回覆率;另外,乳酸具有非常好的生物相容性,這對工業上高產乳酸至關重要;由於乳酸的生物合成已經實現了工業級別的生產,因此,其下遊純化技術也相對成熟。這一切的特徵都使得乳酸成為與1,3-PD聯產的最佳選擇。而事實也證明聯產1,3-PD和L-LAC獲得了非常高的總轉化率(超過90%)。根據現有報導(Biotechnol Bioeng.Metabolism in 1,3-propanediol fed-batch fermentation by a D-lactate deficient mutant of Klebsiella pneumoniae.2009;104(5):965-72.;Bioresour Technol.Production of optically pure d-lactate from glycerol by engineered Klebsiella pneumoniae strain.2014;172:269-75.;Hong A A,Cheng K K et al.Strain isolation and optimization of process parameters for bioconversion of glycerol to lactic acid[J].Journal of chemical technology and biotechnology,2009,84(10):1576-1581.),在克雷伯氏菌中,由甘油直接轉化合成1,3-PD的最高產量為95.4g/L,從甘油到1,3-PD的摩爾轉化率為0.48mol/mol(即48%);從甘油直接轉化L-乳酸的最高產量為85.4g/L,轉化率為0.9mol/mol(即90%),生產強度為0.97g/Lh-1。但是根據現在已有報導,尚無可同時生產1,3-PD和光學純LAC(L-LAC或D-LAC)的菌株。
由於在工業化大規模生產中,必須考慮產物濃度、轉化率,提取成本等因素,如果能夠利用一個菌株,以甘油為底物,以1,3-PD和光學純LAC(L-LAC或D-LAC)為主要產物,高摩爾轉化率的實現同時生產1,3-PD和光學純LAC(L-LAC或D-LAC),將大大降低生產成本,提高生產效率。
2、產酸克雷伯氏菌PLL CCTCC M 2016186易於離心和過濾,有利於生物材料單體1,3-PD和L-LAC的高效生物法生產和提取。
3、產酸克雷伯氏菌PLL CCTCC M 2016186,具有重要的實際應用價值。
4、產酸克雷伯氏菌PLL聯產得到的1,3-PD和L-LAC的濃度差距不大,其質量比都在1:0.1-10範圍內,尤其是在1:0.2-5範圍內,進一步地在1:0.5-2範圍內。
附圖說明
圖1是對產酸克雷伯氏菌(Klebsiella oxytoca)進行基因改造的示意圖。
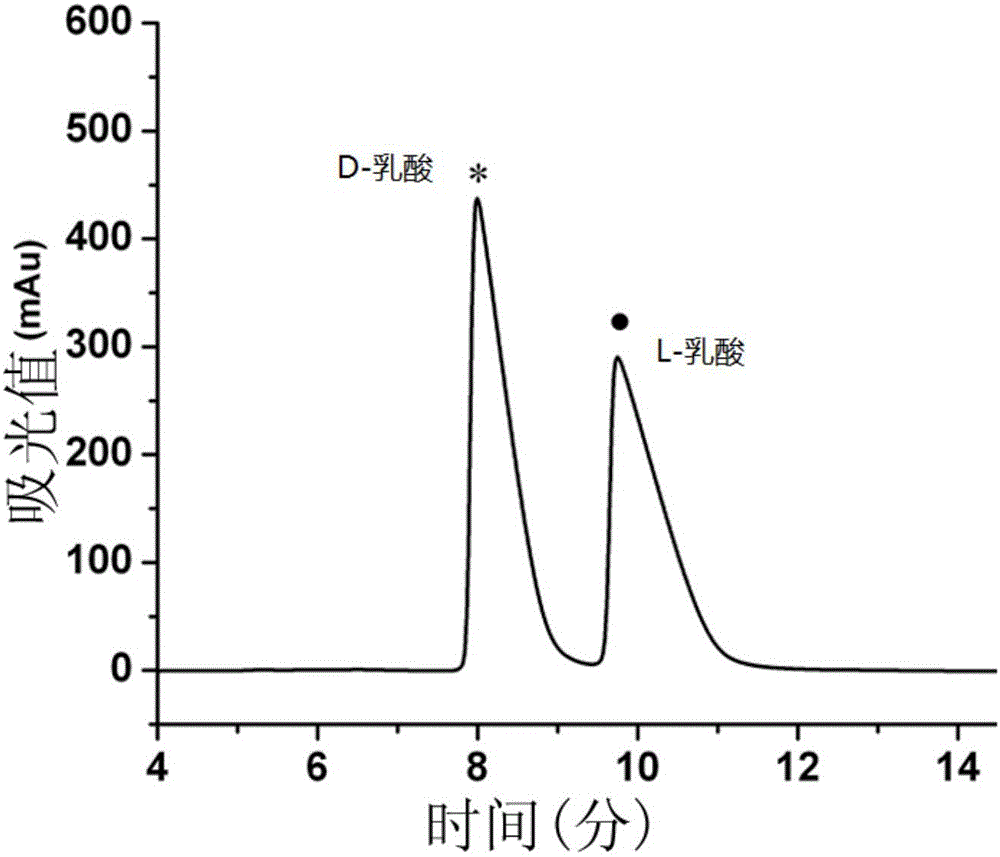
圖2是D-乳酸和L-乳酸的標準品混合物檢測圖。
圖3是產酸克雷伯氏菌PLL CCTCC M 2016186的發酵產物乳酸手性分析檢測圖。
具體實施方式
下面結合實施例對本發明的技術內容做進一步的說明:下述實施例是說明性的,不是限定性的,不能以下述實施例來限定本發明的保護範圍。下述實施例中所使用的實驗方法如無特殊說明,均為常規方法。下述實施例中所用的材料、試劑等,如無特殊說明,均可從商業途徑得到。
本發明所述的基因工程產酸克雷伯氏菌PLL CCTCC M 2016186,由於budA、budB、adhE、ackA-pta、poxB、frdA和dldh基因缺陷,導致α-乙醯乳酸脫羧酶、α-乙醯乳酸合成酶、醛脫氫酶、乙酸激酶、乙醯磷酸轉移酶、丙酮酸氧化酶、富馬酸還原酶和D-乳酸脫氫酶活性丟失,因此2,3-丁二醇、乙醇、乙酸、琥珀酸和D-LAC代謝途徑失活;另外,凝結芽孢桿菌2-6的lldh基因插入到budA基因在基因組中上遊的啟動子之後,使菌株具有了L-乳酸脫氫酶的活性,如圖1所示。從圖1中可以看出,甘油可轉化得到1,3-丙二醇和L-乳酸,L-乳酸脫氫酶催化丙酮酸生成L-乳酸,並且以下代謝途徑失活:α-乙醯乳酸合成酶催化丙酮酸生成α-乙醯乳酸;α-乙醯乳酸脫羧酶催化α-乙醯乳酸生成乙偶姻;丙酮酸氧化酶催化丙酮酸生成乙酸;乙醯磷酸轉移酶催化乙醯輔酶A生成乙醯磷酸;乙醯激酶催化乙醯磷酸生成乙酸;醛脫氫酶催化乙醯輔酶A生成乙醛;富馬酸還原酶催化富馬酸生成琥珀酸;D-乳酸脫氫酶催化丙酮酸生成D-乳酸。在圖1中,差號表示該酶活性丟失,相應的代謝途徑失活。
由於α-乙醯乳酸脫羧酶、α-乙醯乳酸合成酶、醛脫氫酶、乙酸激酶、乙醯磷酸轉移酶、丙酮酸氧化酶、富馬酸還原酶和D-乳酸脫氫酶在產酸克雷伯氏菌中不利於甘油向1,3-PD和L-LAC轉化,因此當這些酶功能缺陷後,則有利於產酸克雷伯氏菌以甘油為底物生產1,3-PD和L-LAC。同時,插入L-乳酸脫氫酶基因到該基因工程產酸克雷伯氏菌中,也有利於丙酮酸向L-LAC轉化,見圖1。本發明正是在這樣的基因改造思路下進行相應的基因工程菌株構建,得到了高產1,3-PD和L-LAC、易純化的上述基因型的基因工程產酸克雷伯氏菌。以下將以具體的菌株對其有益效果進行說明和驗證,但並不表示只有下述具體菌株才具有高產1,3-PD和L-LAC、易純化的有益效果,理論上應該是凡是具備該基因型的產酸克雷伯氏菌均有這樣的有益效果。
如圖2和3所示,使用產酸克雷伯氏菌PLL CCTCC M 2016186,以甘油為底物,可生產高濃度的1,3-PD和高濃度的L-LAC。發酵液中檢測不到D-LAC,因此,生產的L-LAC具有高光學純度(純度大於99.9%)。發酵液中檢測不到2,3-丁二醇、乙醇和甲酸,僅能檢測到少量的乙酸和琥珀酸,發酵液中產物檢測結果如表1所示。
表1:產酸克雷伯氏菌PLL CCTCC M 2016186以甘油為底物發酵液中產物組成
如表1所示,產酸克雷伯氏菌PLL CCTCC M 2016186,以甘油為底物,可生產70.0g/L的1,3-PD和104.0g/L的L-LAC,其中,1,3-PD的摩爾轉化率達到42.5%,L-LAC的摩爾轉化率達到53.4%,兩種主要產物1,3-PD和L-LAC的總轉化率超過90%。發酵液中僅能檢測到少量的副產物。
實施例1:以1,3-PD和LAC為主要產物的菌株的篩選和鑑定
稱取2g土壤樣品加入50mL甘油液體培養基中,置於搖床中37℃培養24小時,搖床轉速為200rpm。然後用無菌的生理鹽水稀釋培養液,分別稀釋10倍、100倍、1000倍和10000倍後塗布到含甘油固體培養基的培養皿中,37℃靜置培養24小時。待長出單菌落後,挑選菌落面積和產酸透明圈面積大的菌落,接種到甘油液體培養基中,置於搖床中37℃培養24小時,搖床轉速為200rpm。對培養液進行離心,測定培養液中1,3-PD和LAC的產量,挑取一株1,3-PD和LAC產量高,同時易於離心的菌株。
將上述菌株多次在甘油固體培養基上劃線分離純化,然後再進行10個循環的培養測試,10次循環培養產生的1,3-PD和LAC產量和轉化率基本保持原有水平,證明上述菌株即是目標菌株,將該菌株命名為PDL-0。測定菌株PDL-0培養液中D-LAC和L-LAC的比例,結果表明,菌株PDL-0生產的LAC中,D-LAC的比例大於99.9%,L-LAC的比例小於0.01%。
抽提菌株PDL-0的全基因組,然後PCR擴增菌株PDL-0的16S rRNA的基因序列,將PCR產物測序,測序得到的16S rRNA基因序列如SEQ ID NO:9所示。菌株PDL-0的16S rRNA基因序列與NCBI資料庫(http://www.ncbi.nlm.nih.gov/)中其他產酸克雷伯氏菌的16S rRNA基因序列具有99%的同源性,分析結果表明,菌株PDL-0為產酸克雷伯氏菌(Klebsiella oxytoca)。
其中,所述甘油液體培養基的配方為:酵母粉3g/L、K2HPO4·3H2O 10g/L、KH2PO42g/L、NH4Cl 1g/L、MgSO4·7H2O 0.5g/L、FeCl3·6H2O 20mg/L、CoCl2·6H2O 50mg/L和甘油20g/L;121℃滅菌20分鐘。
其中,所述甘油固體培養基的配方為:酵母粉3g/L、K2HPO4·3H2O 10g/L、KH2PO42g/L、NH4Cl 1g/L、MgSO4·7H2O 0.5g/L、FeCl3·6H2O 20mg/L、CoCl2·6H2O 50mg/L,甘油20g/L和瓊脂粉15g/L;121℃滅菌20分鐘。
實施例2:產酸克雷伯氏菌PDL-0中α-乙醯乳酸脫羧酶基因(budA)和α-乙醯乳酸合成酶基因(budB)的缺陷
(1)用於在產酸克雷伯氏菌PDL-0中部分缺失budA基因的載體的構建
根據budA基因序列(如SEQ ID NO:1所示)設計引物,PCR擴增budA基因的上遊和下遊同源片段。以產酸克雷伯氏菌PDL-0的基因組DNA為模板,使用引物budA-1:5』-ACATGATTACGAATTCATGAACCATTCTGCTGAATG-3』(如SEQ ID NO:10所示)和引物budA-2:5』-AACGGGCTGGCATCACCGCGAAGGGCGTGC-3』(如SEQ ID NO:11所示)進行PCR擴增,得到上遊同源片段;使用引物budA-3:5』-CGCGGTGATGCCAGCCCGTTTTCCGCTTCA-3』(如SEQ ID NO:12所示)和引物budA-4:5』-TACCGAGCTCGAATTCTTAGTTTTCGACTGAGCGAA-3』(如SEQ ID NO:13所示)進行PCR擴增,得到下遊同源片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到上遊和下遊同源片段。
使用限制性內切酶EcoRI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K。自殺質粒pKR6K可通過將質粒pK18mobsacB(優寶生物公司)的複製子替換成質粒pCAM140的複製子獲得。質粒pCAM140的複製子的序列可通過基因合成的方式獲得,質粒pCAM140的序列見文獻報導(Wilson K J,Sessitsch A,Corbo J C,et al.β-Glucuronidase(GUS)transposons for ecological and genetic studies of rhizobia and other Gram-negative bacteria[J].Microbiology,1995,141(7):1691-1705.)。
使用無縫克隆和組裝試劑盒(pEASY-Uni Seamless Cloning and Assembly Kit,北京全式金生物技術有限公司),將上遊同源片段、下遊同源片段和線性化的質粒pKR6K連接,得到可部分缺失budA基因的自殺質粒pKR6K-ΔbudA。
(2)budA基因部分缺失的產酸克雷伯氏菌的構建
將pKR6K-ΔbudA轉化至大腸桿菌S17-1(λpir)(北京全式金生物技術有限公司)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔbudA)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔbudA)與受體菌產酸克雷伯氏菌PDL-0進行雙親本雜交,使pKR6K-ΔbudA上的budA基因上遊同源片段和下遊同源片段與產酸克雷伯氏菌PDL-0的基因組發生同源重組,從而使產酸克雷伯氏菌PDL-0的budA基因缺失200bp,達到使budA基因缺陷的目的,具體方法為:
a.分別接種活化後的供體菌和受體菌至5mL LB液體培養基中,37℃搖床中,200rpm培養2-3小時,當供體菌和受體菌同時生長到OD620nm為0.5-0.8;將5mL供體菌菌液離心,無菌生理鹽水洗兩遍;將1mL受體菌菌液離心,無菌生理鹽水洗兩遍;將上述供體菌和受體菌的菌體一共用100μL無菌生理鹽水重懸,將重懸液全部滴在LB固體培養基平板中間,平板正面放置,37℃培養12-18小時。
b.將步驟a中LB固體培養基平板上的菌落用無菌生理鹽水和刮刀刮下,無菌生理鹽水洗兩遍,進行適當稀釋,塗布於加入了50μg/mL卡那黴素的M9固體培養基平板,37℃培養24-36小時。
c.挑取步驟b中M9固體培養基平板上生長的單菌落至加入50μg/mL卡那黴素的5mL LB液體培養基中,37℃,200rpm培養12小時。轉接菌液至新鮮的5mL LB液體培養基中(不添加卡那黴素),37℃,200rpm培養12小時。
d.將上述菌液適當稀釋,塗布於LAS固體培養基平板,25℃培養24小時。
e.挑取步驟d中LAS固體培養基平板上生長的單菌落至5mL LB液體培養基中,37℃,200rpm培養12小時,提取基因組DNA,使用引物budA-1:5』-ACATGATTACGAATTCATGAACCATTCTGCTGAATG-3』(如SEQ ID NO:10所示)和引物budA-4:5』-TACCGAGCTCGAATTCTTAGTTTTCGACTGAGCGAA-3』(如SEQ ID NO:13所示)進行PCR驗證。獲得的budA基因缺陷的菌株。
其中,所述LB液體培養基配方為:蛋白腖10g/L、酵母粉5g/L和NaCl10g/L,121℃滅菌20分鐘。
其中,所述LB固體培養基配方為:蛋白腖10g/L、酵母粉5g/L、NaCl 10g/L和瓊脂粉15g/L,121℃滅菌20分鐘。
其中,所述M9固體培養基配方為:Na2HPO4·12H2O 1.7g/L、KH2PO40.3g/L、NaCl 0.05g/L、NH4Cl 0.1g/L、檸檬酸三鈉0.5g/L和瓊脂粉15g/L,121℃滅菌20分鐘。
其中,所述LAS固體培養基配方為:蛋白腖10g/L、酵母粉5g/L、蔗糖150g/L和瓊脂粉15g/L,115℃滅菌20分鐘。
(3)用於在budA基因缺陷的菌株中部分缺失budB基因的載體的構建
根據budB基因序列(如SEQ ID NO:2所示)設計引物,PCR擴增budB基因的上遊和下遊同源片段。以產酸克雷伯氏菌PDL-0的基因組DNA為模板,使用引物budB-1:5』-ACGCGAATTCGTGGATAATCAACATCAACCGCGCC-3』(如SEQ ID NO:14所示)和引物budB-2:5』-ACGCGGATCCGGGGCGTCCCTGCTCGGC-3』(如SEQ ID NO:15所示)進行PCR擴增,得到上遊同源片段;使用引物budB-3:5』-ACGCGGATCCATCGCCCGCTATCTCTACAGCTTCC-3』(如SEQ ID NO:16所示)和引物budB-4:5』-ACGCCTGCAGATTTGACTGAGATGAAGCTGGCCCA-3』(如SEQ ID NO:17所示)進行PCR擴增,得到下遊同源片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到上遊和下遊同源片段。
使用限制性內切酶EcoRI和BamHI酶切上遊同源片段,使用限制性內切酶BamHI和PstI酶切下遊同源片段,使用限制性內切酶EcoRI和PstI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K和帶有粘性末端的上遊和下遊同源臂片段。
使用T4連接酶(NEB公司)將帶有粘性末端的上遊同源片段、下遊同源片段和線性化的質粒pKR6K連接,得到可部分缺失budB基因的自殺質粒pKR6K-ΔbudB。
(4)budB基因部分缺失的產酸克雷伯氏菌的構建
將pKR6K-ΔbudB轉化至大腸桿菌S17-1(λpir)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔbudB)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔbudB)與受體菌budA基因缺陷的菌株進行雙親本雜交,使pKR6K-ΔbudB上的budB基因上遊同源片段和下遊同源片段與budA基因缺陷的菌株的基因組發生同源重組,從而budA基因缺陷的菌株的budB基因缺失722bp,達到使budB基因缺陷的目的。具體方法與步驟(2)相同,只是PCR驗證時,使用引物budB-1:5』-ACGCGAATTCGTGGATAATCAACATCAACCGCGCC-3』(如SEQ ID NO:14所示)和引物budB-4:5』-ACGCCTGCAGATTTGACTGAGATGAAGCTGGCCCA-3』(如SEQ ID NO:17所示)進行PCR驗證。獲得的budA基因和budB基因缺陷的菌株,命名為產酸克雷伯氏菌PDL-1。
實施例3:產酸克雷伯氏菌PDL-1中醛脫氫酶基因(adhE)的缺陷
(1)用於在產酸克雷伯氏菌PDL-1中部分缺失adhE基因的載體的構建
根據adhE基因序列(如SEQ ID NO:3所示)設計引物,PCR擴增adhE基因的上遊和下遊同源片段。以產酸克雷伯氏菌PDL-1的基因組DNA為模板,使用引物adhE-1:5』-ACATGATTACGAATTCATGGCTGTTACTAATGTCGC-3』(如SEQ ID NO:18所示)和引物adhE-2:5』-TGCTGTCTGTTGGCGTTACGGGTCTTCAGG-3』(如SEQ ID NO:19所示)進行PCR擴增,得到上遊同源片段;使用引物adhE-3:5』-CGTAACGCCAACAGACAGCATTCAGCCAGT-3』(如SEQ ID NO:20所示)和引物adhE-4:5』-TACCGAGCTCGAATTCTTAAGCGGATTTTTTCGCTT-3』(如SEQ ID NO:21所示)進行PCR擴增,得到下遊同源片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到上遊和下遊同源片段。
使用限制性內切酶EcoRI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K。
使用無縫克隆和組裝試劑盒(pEASY-Uni Seamless Cloning and Assembly Kit,北京全式金生物技術有限公司),將上遊同源片段、下遊同源片段和線性化的質粒pKR6K連接,得到可部分缺失adhE基因的自殺質粒pKR6K-ΔadhE。
(2)adhE基因部分缺失的產酸克雷伯氏菌的構建
將pKR6K-ΔadhE轉化至大腸桿菌S17-1(λpir)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔadhE)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔadhE)與受體菌產酸克雷伯氏菌PDL-1進行雙親本雜交,使pKR6K-ΔadhE上的adhE基因上遊同源片段和下遊同源片段與產酸克雷伯氏菌PDL-1的基因組發生同源重組,從而使產酸克雷伯氏菌PDL-1的adhE基因缺失1876bp,達到使adhE基因缺陷的目的。具體方法與實施例2相同,只是PCR驗證時,使用引物adhE-1:5』-ACATGATTACGAATTCATGGCTGTTACTAATGTCGC-3』(如SEQ ID NO:18所示)和引物adhE-4:5』-TACCGAGCTCGAATTCTTAAGCGGATTTTTTCGCTT-3』(如SEQ ID NO:21所示)進行PCR驗證。獲得的adhE基因缺陷的菌株,命名為產酸克雷伯氏菌PDL-2。
實施例4:產酸克雷伯氏菌PDL-2中乙酸激酶和乙醯磷酸轉移酶基因(ackA-pta)的缺陷
(1)用於在產酸克雷伯氏菌PDL-2中部分缺失ackA-pta基因的載體的構建
根據ackA-pta基因序列(如SEQ ID NO:4所示)設計引物,PCR擴增ackA-pta基因的上遊和下遊同源片段。以產酸克雷伯氏菌PDL-2的基因組DNA為模板,使用引物ackA-pta-1:5』-ACATGATTACGAATTCATGTCGAGTAAGTTAGTACT-3』(如SEQ ID NO:22所示)和引物ackA-pta-2:5』-CACGCGCGGTCCTCAGCGATACCGATCAGG-3』(如SEQ ID NO:23所示)進行PCR擴增,得到上遊同源片段;使用引物ackA-pta-3:5』-ATCGCTGAGGACCGCGCGTGGCCATGCTCT-3』(如SEQ ID NO:24所示)和引物ackA-pta-4:5』-TACCGAGCTCGAATTCTTATGCTTGCTGCTGGGACG-3』(如SEQ ID NO:25所示)進行PCR擴增,得到下遊同源片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到上遊和下遊同源片段。
使用限制性內切酶EcoRI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K。
使用無縫克隆和組裝試劑盒(pEASY-Uni Seamless Cloning and Assembly Kit,北京全式金生物技術有限公司),將上遊同源片段、下遊同源片段和線性化的質粒pKR6K連接,得到可部分缺失ackA-pta基因的自殺質粒pKR6K-ΔackA-pta。
(2)ackA-pta基因部分缺失的產酸克雷伯氏菌的構建
將pKR6K-ΔackA-pta轉化至大腸桿菌S17-1(λpir)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔackA-pta)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔackA-pta)與受體菌產酸克雷伯氏菌PDL-2進行雙親本雜交,使pKR6K-ΔackA-pta上的ackA-pta基因上遊同源片段和下遊同源片段與產酸克雷伯氏菌PDL-2的基因組發生同源重組,從而使產酸克雷伯氏菌PDL-2的ackA-pta基因缺失2749bp,達到使ackA-pta基因缺陷的目的。具體方法與實施例2相同,只是PCR驗證時,使用引物ackA-pta-1:5』-ACATGATTACGAATTCATGTCGAGTAAGTTAGTACT-3』(如SEQ ID NO:22所示)和引物ackA-pta-4:5』-TACCGAGCTCGAATTCTTATGCTTGCTGCTGGGACG-3』(如SEQ ID NO:25所示)進行PCR驗證。獲得的ackA-pta基因缺陷的菌株,命名為產酸克雷伯氏菌PDL-3。
實施例5:產酸克雷伯氏菌PDL-3中丙酮酸氧化酶基因(poxB)的缺陷
(1)用於在產酸克雷伯氏菌PDL-3中部分缺失poxB基因的載體的構建
根據poxB基因序列(如SEQ ID NO:5所示)設計引物,PCR擴增poxB基因的上遊和下遊同源片段。以產酸克雷伯氏菌PDL-3的基因組DNA為模板,使用引物poxB-1:5』-ACATGATTACGAATTCATGAAACAGACCGTGGCGGC-3』(如SEQ ID NO:26所示)和引物poxB-2:5』-AAAATCCCCCGGGTTGAGACCAGTTCACAG-3』(如SEQ ID NO:27所示)進行PCR擴增,得到上遊同源片段;使用引物poxB-3:5』-GTCTCAACCCGGGGGATTTTCTCTCGCTGG-3』(如SEQ ID NO:28所示)和引物poxB-4:5』-TACCGAGCTCGAATTCTTACCTTAGCCAGTTAGTTT-3』(如SEQ ID NO:29所示)進行PCR擴增,得到下遊同源片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到上遊和下遊同源片段。
使用限制性內切酶EcoRI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K。
使用無縫克隆和組裝試劑盒(pEASY-Uni Seamless Cloning and Assembly Kit,北京全式金生物技術有限公司),將上遊同源片段、下遊同源片段和線性化的質粒pKR6K連接,得到可部分缺失poxB基因的自殺質粒pKR6K-ΔpoxB。
(2)poxB基因部分缺失的產酸克雷伯氏菌的構建
將pKR6K-ΔpoxB轉化至大腸桿菌S17-1(λpir)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔpoxB)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔpoxB)與受體菌產酸克雷伯氏菌PDL-3進行雙親本雜交,使pKR6K-ΔpoxB上的poxB基因上遊同源片段和下遊同源片段與產酸克雷伯氏菌PDL-3的基因組發生同源重組,從而使產酸克雷伯氏菌PDL-3的poxB基因缺失919bp,達到使poxB基因缺陷的目的。具體方法與實施例2相同,只是PCR驗證時,使用引物poxB-1:5』-ACATGATTACGAATTCATGAAACAGACCGTGGCGGC-3』(如SEQ ID NO:26所示)和引物poxB-4:5』-TACCGAGCTCGAATTCTTACCTTAGCCAGTTAGTTT-3』(如SEQ ID NO:29所示)進行PCR驗證。獲得的poxB基因缺陷的菌株,命名為產酸克雷伯氏菌PDL-4。
實施例6:產酸克雷伯氏菌PDL-4中富馬酸還原酶基因(frdA)的缺陷
(1)用於在產酸克雷伯氏菌PDL-4中部分缺失frdA基因的載體的構建
根據frdA基因序列(如SEQ ID NO:6所示)設計引物,PCR擴增frdA基因的上遊和下遊同源片段。以產酸克雷伯氏菌PDL-4的基因組DNA為模板,使用引物frdA-1:5』-ACATGATTACGAATTCGTGCAAACTTTTCAAGCCGA-3』(如SEQ ID NO:30所示)和引物frdA-2:5』-GTAGATGCCGAGCCGGTTTTATCGGCAGCG-3』(如SEQ ID NO:31所示)進行PCR擴增,得到上遊同源片段;使用引物frdA-3:5』-AAAACCGGCTCGGCATCTACCGTACGCCGG-3』(如SEQ ID NO:32所示)和引物frdA-4:5』-TACCGAGCTCGAATTCTCAGCCATTCGTCGTCTCCT-3』(如SEQ ID NO:33所示)進行PCR擴增,得到下遊同源片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到上遊和下遊同源片段。
使用限制性內切酶EcoRI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K。
使用無縫克隆和組裝試劑盒(pEASY-Uni Seamless Cloning and Assembly Kit,北京全式金生物技術有限公司),將上遊同源片段、下遊同源片段和線性化的質粒pKR6K連接,得到可部分缺失frdA基因的自殺質粒pKR6K-ΔfrdA。
(2)frdA基因部分缺失的產酸克雷伯氏菌的構建
將pKR6K-ΔfrdA轉化至大腸桿菌S17-1(λpir)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔfrdA)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔfrdA)與受體菌產酸克雷伯氏菌PDL-4進行雙親本雜交,使pKR6K-ΔfrdA上的frdA基因上遊同源片段和下遊同源片段與產酸克雷伯氏菌PDL-4的基因組發生同源重組,從而使產酸克雷伯氏菌PDL-4的frdA基因缺失991bp,達到使frdA基因缺陷的目的。具體方法與實施例2相同,只是PCR驗證時,使用引物frdA-1:5』-ACATGATTACGAATTCGTGCAAACTTTTCAAGCCGA-3』(如SEQ ID NO:30所示)和引物frdA-4:5』-TACCGAGCTCGAATTCTCAGCCATTCGTCGTCTCCT-3』(如SEQ ID NO:33所示)進行PCR驗證。獲得的frdA基因缺陷的菌株,命名為產酸克雷伯氏菌PDL-5。
實施例7:產酸克雷伯氏菌PDL-5中D-乳酸脫氫酶基因(dldh)的缺陷
(1)用於在產酸克雷伯氏菌PDL-5中缺失dldh基因的載體的構建
根據dldh基因序列(如SEQ ID NO:7所示)設計引物,PCR擴增dldh基因的上遊和下遊同源片段。以產酸克雷伯氏菌PDL-5的基因組DNA為模板,使用引物dldh-1:5』-ACATGATTACGAATTCATGGAGCATCTGCACATGAA-3』(如SEQ ID NO:34所示)和引物dldh-2:5』-AAAGGGAAAGGAATAAAGACTTTTCTCCAGTGATAATACC-3』(如SEQ ID NO:35所示)進行PCR擴增,得到上遊同源片段;使用引物dldh-3:5』-CTGGAGAAAAGTCTTTATTCCTTTCCCTTTTGTGCTC-3』(如SEQ ID NO:36所示)和引物dldh-4:5』-TACCGAGCTCGAATTCTTATCAGAACTGATCTTCTT-3』(如SEQ ID NO:37所示)進行PCR擴增,得到下遊同源片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到上遊和下遊同源片段。
使用限制性內切酶EcoRI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K。
使用無縫克隆和組裝試劑盒(pEASY-Uni Seamless Cloning and Assembly Kit,北京全式金生物技術有限公司),將上遊同源片段、下遊同源片段和線性化的質粒pKR6K連接,得到可缺失dldh基因的自殺質粒pKR6K-Δdldh。
(2)dldh基因缺失的產酸克雷伯氏菌的構建
將pKR6K-Δdldh轉化至大腸桿菌S17-1(λpir)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-Δdldh)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-Δdldh)與受體菌產酸克雷伯氏菌PDL-5進行雙親本雜交,使pKR6K-Δdldh上的dldh基因上遊同源片段和下遊同源片段與產酸克雷伯氏菌PDL-5的基因組發生同源重組,從而使產酸克雷伯氏菌PDL-5的dldh基因缺失990bp,達到dldh基因缺陷的目的。具體方法與實施例2相同,只是PCR驗證時,使用引物dldh-1:5』-ACATGATTACGAATTCATGGAGCATCTGCACATGAA-3』(如SEQ ID NO:34所示)和引物dldh-4:5』-TACCGAGCTCGAATTCTTATCAGAACTGATCTTCTT-3』(如SEQ ID NO:37所示)進行PCR驗證。獲得的dldh基因缺陷的菌株,命名為產酸克雷伯氏菌PDL-6。
實施例2-7給出了構建budA、budB、adhE、ackA-pta、poxB、frdA和dldh酶失活菌株的方法,即通過基因同源重組的方式來實現基因缺陷從而導致其所編碼的酶失活。然而導致酶失活的方式並不限於基因同意重組,還可以是:小RNA幹擾,點突變,加入相關酶的抑制劑等等。
實施例8:將凝結芽孢桿菌2-6的L-乳酸脫氫酶基因(lldh)插入產酸克雷伯氏菌PDL-6的α-乙醯乳酸脫羧酶基因(budA)在基因組中上遊的啟動子之後
(1)用於在產酸克雷伯氏菌PDL-6的budA基因在基因組中上遊的啟動子之後插入凝結芽孢桿菌2-6的lldh基因的載體的構建
根據產酸克雷伯氏菌基因組序列設計引物,PCR擴增budA基因在基因組上遊和下遊的同源片段。以產酸克雷伯氏菌PDL-6的基因組DNA為模板,使用引物budA-lldh-1:5』-ACATGATTACGAATTCATTGCCCTCGACCTGATGTAAC-3』(如SEQ ID NO:38所示)和引物budA-lldh-2:5』-ATTGACTTTTTTCATTACCCGCTTCCTCGTTCAAC-3』(如SEQ ID NO:39所示)進行PCR擴增,得到上遊同源片段;使用引物budA-lldh-3:5』-GCACCGATATTGTAAGTCACTACAGAAGGAATCCAGCAAT-3』(如SEQ ID NO:40所示)和引物budA-lldh-4:5』-TACCGAGCTCGAATTCGCACGGCAAACTTACTTGAGCTA-3』(如SEQ ID NO:41所示)進行PCR擴增,得到下遊同源片段。根據凝結芽孢桿菌2-6的lldh基因序列(如SEQ ID NO:8所示)設計引物,PCR擴增凝結芽孢桿菌2-6的lldh基因。以凝結芽孢桿菌2-6的基因組DNA為模板,使用引物lldh-1:5』-ACGAGGAAGCGGGTAATGAAAAAAGTCAATCGTATTGCA-3』(如SEQ ID NO:42所示)和引物lldh-2:5』-TCCTTCTGTAGTGACTTACAATATCGGTGCCATTGTTT-3』(如SEQ ID NO:43所示)進行PCR擴增,得到凝結芽孢桿菌2-6的lldh基因片段。PCR擴增條件為:95℃5分鐘;95℃30秒,60℃30秒,72℃1分鐘,共30個循環;72℃5分鐘。PCR反應結束後,將PCR擴增產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到budA基因在基因組上遊和下遊的同源片段,以及凝結芽孢桿菌2-6的lldh基因片段。
使用限制性內切酶EcoRI酶切自殺質粒pKR6K(Wang et al.,J.Biol.Chem.2014,289:6080-6090),將酶切產物進行1.0%瓊脂糖凝膠電泳,回收並純化,得到線性化的質粒pKR6K。
使用無縫克隆和組裝試劑盒(pEASY-Uni Seamless Cloning and Assembly Kit,北京全式金生物技術有限公司),將budA基因在基因組上遊和下遊的同源片段,以及凝結芽孢桿菌2-6的lldh基因片段與線性化的質粒pKR6K連接,得到用於在產酸克雷伯氏菌PDL-6中替換budB基因為凝結芽孢桿菌2-6的lldh基因的載體pKR6K-ΔbudA::lldh。
(2)攜帶凝結芽孢桿菌2-6的lldh基因的產酸克雷伯氏菌的構建
將pKR6K-ΔbudA::lldh轉化至大腸桿菌S17-1(λpir)中,得到供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔbudA::lldh)。將供體菌大腸桿菌S17-1(λpir)(pKR6K-ΔbudA::lldh)與受體菌產酸克雷伯氏菌PDL-6進行雙親本雜交,使pKR6K-ΔbudA::lldh上的budA基因上遊同源片段和下遊同源片段與產酸克雷伯氏菌PDL-6的基因組發生同源重組,從而將凝結芽孢桿菌2-6的lldh基因插入到產酸克雷伯氏菌PDL-6的budA基因在基因組中上遊的啟動子之後。具體方法與實施例2相同,只是PCR驗證時,使用引物budA-lldh-1:5』-ACATGATTACGAATTCATTGCCCTCGACCTGATGTAAC-3』(如SEQ ID NO:38所示)和budA-lldh-4:5』-TACCGAGCTCGAATTCGCACGGCAAACTTACTTGAGCTA-3』(如SEQ ID NO:41所示)進行PCR驗證。獲得的攜帶凝結芽孢桿菌2-6的lldh基因的菌株,命名為產酸克雷伯氏菌PLL CCTCC M 2016186。
實施例9:使用產酸克雷伯氏菌PLL CCTCC M 2016186分批發酵生產1,3-PD和L-LAC
(1)菌株選擇:選用產酸克雷伯氏菌PLL CCTCC M 2016186;
(2)種子培養:選用步驟(1)的菌株,在無菌條件下接種至甘油培養基中,培養溫度為30℃,搖床震蕩轉速為200rpm,培養時間為15小時,製得種子培養液;
(3)發酵:將步驟(2)中製得的種子培養液接種至裝有甘油培養基的發酵罐中,接種量為5%(v/v),發酵溫度為30℃,通氣量為0.5vvm,攪拌轉速為200rpm,發酵過程中,使用25%(w/v)氫氧化鈉水溶液作為中和劑調節發酵液pH為7.0,發酵方式為分批發酵,當甘油培養基中的甘油耗盡時,停止發酵。
其中,所述甘油培養基的配方為:酵母粉2g/L、K2HPO4·3H2O 5g/L、KH2PO41g/L、NH4Cl 2g/L、MgSO4·7H2O 0.1g/L、FeCl3·6H2O 10mg/L、CoCl2·6H2O 10mg/L和甘油60g/L;121℃滅菌20分鐘。
發酵14小時後,甘油培養基中的甘油耗盡,停止發酵,檢測發酵液中的產物組成和濃度。發酵的主要產物為1,3-PD和L-LAC,1,3-PD濃度為22.7g/L,L-LAC濃度為32.4g/L。副產物僅能檢測到少量的乙酸、甲酸和琥珀酸,乙酸濃度為0.7g/L,琥珀酸濃度為0.3g/L,丙酮酸濃度為0.4g/L。發酵液中檢測不到2,3-丁二醇、乙醇和甲酸。1,3-PD的摩爾轉化率為45.8%,L-lac的摩爾轉化率為50.0%。兩種產物的總轉化率為95.8%。
實施例10:使用產酸克雷伯氏菌PLL CCTCC M 2016186分批補料發酵生產1,3-PD和L-LAC
(1)菌株選擇:選用產酸克雷伯氏菌PLL CCTCC M 2016186;
(2)種子培養:選用步驟(1)的菌株,在無菌條件下接種至甘油培養基中,培養溫度為37℃,搖床震蕩轉速為150rpm,培養時間為12小時,製得種子培養液;
(3)發酵:將步驟(2)中製得的種子培養液接種至裝有甘油培養基的發酵罐中,接種量為2.5%(v/v),發酵溫度為37℃,通氣量為1vvm,攪拌轉速為250rpm,發酵過程中,使用25%(w/v)的氫氧化鈣和水的混合乳液作為中和劑調節發酵液pH為6.5,發酵方式為分批補料發酵,當甘油培養基中的甘油耗盡時,通過向發酵罐中補加700g/L的甘油溶液,控制發酵液中甘油濃度為5-30g/L,當發酵液中1,3-PD或L-LAC濃度不再升高時,停止發酵。
其中,所述甘油培養基的配方為:酵母粉5g/L、K2HPO4·3H2O 10g/L、KH2PO42g/L、NH4Cl 1g/L、MgSO4·7H2O 0.1g/L、FeCl3·6H2O 20mg/L、CoCl2·6H2O 15mg/L和甘油20g/L;121℃滅菌20分鐘。
發酵30小時後,發酵液中1,3-PD和L-LAC濃度不再升高時,停止發酵,檢測發酵液中的產物組成和濃度。發酵的主要產物為1,3-PD和L-LAC,1,3-PD濃度為70.0g/L,L-LAC濃度為104.0g/L。副產物僅能檢測到少量的乙酸和琥珀酸,乙酸濃度為1.5g/L,琥珀酸濃度為0.7g/L。發酵液中檢測不到2,3-丁二醇、乙醇、甲酸等其他副產物。1,3-PD的摩爾轉化率為42.5%,L-乳酸的摩爾轉化率為53.4%。兩種產物的總轉化率為95.9%。
實施例9和10隻是本發明在應用方法上的兩個較佳實施方式,其所限定的數值在合理範圍內的變化也可以實現同一目的,因此不能以實施例9和10中給出的數值對本發明進行限制。雖然實施例9和10是對產酸克雷伯氏菌PLL的發酵產物進行檢測,證明了產酸克雷伯氏菌PLL可以生產高摩爾轉化率的1,3-PD和L-LAC,但是並不能僅僅理解為本發明所公開的產酸克雷伯氏菌進行基因改造的技術方案只適用於產酸克雷伯氏菌PLL,而應該理解為只要是通過本發明所公開的對產酸克雷伯氏菌進行基因改造的技術方案所得到的人工菌都具有同時提高1,3-PD和L-LAC的摩爾轉化率和減少副產物的能力。此外,實施例1-10是以產酸克雷伯氏菌PDL-0為野生菌進行基因改造的,若通過其他與產酸克雷伯氏菌PDL-0具有相同代謝通路的菌進行同樣的基因改造,應當理解為也能同時提高1,3-PD和L-LAC的摩爾轉化率和減少副產物。
使用產酸克雷伯氏菌PLL CCTCC M 2016186,以甘油為底物,進行分批補料發酵,得到的發酵液中目標產物1,3-PD和L-LAC濃度高,副產物種類少且濃度低,此外,產酸克雷伯氏菌PLL CCTCC M 2016186易於離心和過濾,這些優勢都有利於1,3-PD和L-LAC的高效生物法生產,同時有利於產物的提取,說明本發明的產酸克雷伯氏菌PLL CCTCC M 2016186,具有重要的實際應用價值。
實施例11:外源引入1,3-PD合成途徑
(1)菌株選擇:選用大腸桿菌K12;
(2)基因工程改造,在K12中引入外源1,3-PD合成途徑
選用產酸克雷伯氏菌中的1,3-PD合成途徑中的甘油脫水酶編碼基因dhaB及1,3-PD氧化還原酶編碼基因dhaT,進行PCR克隆後連接到質粒DNA pet-Duet上,並轉化到大腸桿菌K12中。該菌確定為K12-dhaBdhaT。
以上詳細描述了本發明的較佳具體實施例。應當理解,本領域的普通技術無需創造性勞動就可以根據本發明的構思作出諸多修改和變化。因此,凡本技術領域中技術人員依本發明的構思在現有技術的基礎上通過邏輯分析、推理或者有限的實驗可以得到的技術方案,皆應在由權利要求書所確定的保護範圍內。