孔隙型微米管結構催化儲氫協同功效材料的製備方法與流程
2023-12-05 23:42:31 3
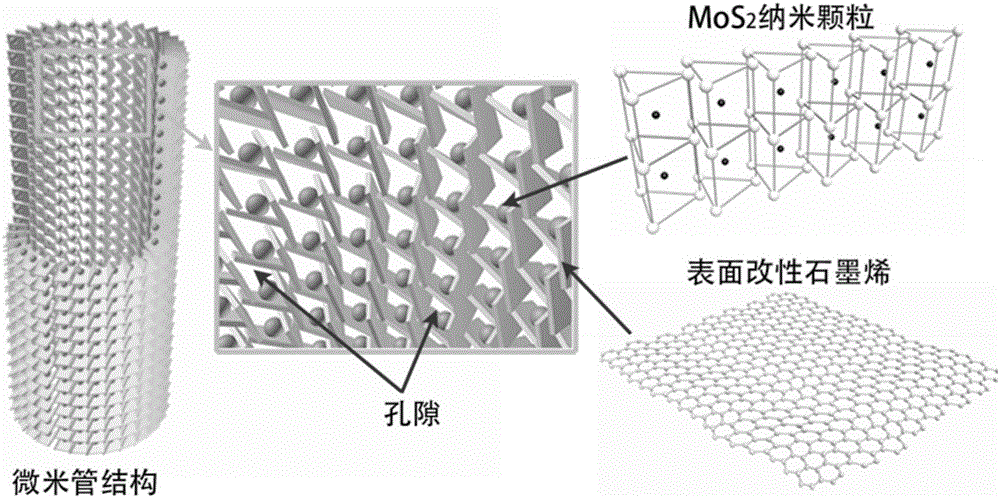
本發明涉及電解水製備氫氣催化和儲氫材料領域,特別是一種孔隙型微米管結構催化儲氫協同功效材料的製備方法。
背景技術:
隨著經濟社會的發展,石油、煤炭等不可再生能源已不能滿足人類的需要。氫氣的燃燒熱值高,燃燒產物無汙染,可循環利用,因而是最有潛力的新能源。
實現氫能源大規模廣泛應用的關鍵在於廉價而又高效的制氫技術和安全高效的儲氫技術的研發。通過電解水工業化生產製備氫氣是大規模獲得潔淨、可持續能源氫燃料的有效手段,高效制氫電極催化材料和高效儲氫材料的研究是氫燃料製備和儲存技術得以突破的關鍵所在。迄今為止,已研發的氫催化材料和儲氫材料都局限於各自的氫製備領域和儲氫技術領域,導致氫製備技術和儲氫技術分離,難以實現氫製備和儲存一體化技術的發展,從而制約了氫能源大規模廣泛應用。
技術實現要素:
為了克服現有技術的不足,本發明提供了一種孔隙型微米管結構催化儲氫協同功效材料的製備方法,根據本發明方法製備的催化儲氫協同功效材料,是由石墨烯網絡構建的微米管結構材料,具有中空結構,微米管壁具有連通孔隙,MoS2納米顆粒固定於微米管壁石墨烯網絡內。這種孔隙型微米管結構催化儲氫協同功效材料既具有對電解水製備氫氣的催化效應,也具有高效吸附儲氫功能。
本發明解決其技術問題所採用的技術方案是:一種孔隙型微米管結構催化儲氫協同功效材料的製備方法,包括按順序進行的如下步驟:
步驟[1]石墨烯表面羧基和羥基基團改性;
步驟[2]石墨烯網狀聯接;
步驟[3]MoS2納米顆粒在空間網狀石墨烯上液相沉降固定及孔隙型微米管的搭建。
所述步驟[1]具體包括:
a1.石墨烯表面羧基基團改性:將石墨烯、分析純的正已酸酐、鄰苯二甲酸酐加入到乙烯乙二醇醚中,形成羧基改性液,超聲波攪拌20-40分鐘,在溫度為30-70℃環境下恆溫反應0.5-1.5小時,冷卻至室溫,過濾固相物質,用丙酮洗滌,在乾燥箱中40-60℃下乾燥8-12小時;
a2.石墨烯表面羥基基團改性:將石墨烯、分析純的磷酸二氫鉀、氫氧化鋇和乙酸亞鐵加入去離子水中,形成羥基改性液,超聲波攪拌30-40分鐘,在室溫下反應1-1.5小時,過濾固相物質,用去離子水洗滌,在乾燥箱中50-60℃下乾燥10-13小時。
優選的,步驟a1中所述羧基改性液中石墨烯的濃度為60g/L-110g/L、正已酸酐的濃度為160mL/L-200mLg/L、鄰苯二甲酸酐的濃度為40g/L-60g/L。
優選的,步驟a2中羥基改性液中的石墨烯濃度為100g/L-130g/L、磷酸二氫鉀濃度為20g/L-65g/L、氫氧化鋇濃度為10g/L-20g/L、乙酸亞鐵濃度為30g/L-45g/L。
所述步驟[2]具體為:
將表面羧基基團和羥基基團改性的石墨烯各100-120g、分析純的三苯基磷-多滷代甲烷20-40g和二環已基碳二亞胺5-10g加入600-800mL的乙二醇單甲醚中,超聲波攪拌5-7分鐘,在室溫下反應20-35分鐘,過濾固相物質,用丙酮洗滌後再以去離子水清洗,在乾燥箱中40-60℃下乾燥16-18小時,得網狀連接石墨烯。
所述步驟[3]具體包括:
b1.MoS2納米顆粒在空間網狀石墨烯上沉降固定:將網狀聯接石墨烯、MoS2納米顆粒、氧化鋅微米棒、分析純的葡萄糖、鉬酸鈉和磷酸二氫銨加入去離子水形成沉降液;超聲波攪拌1-1.5小時,待沉降液固體沉澱後,過濾得沉積物。
b2.孔隙型微米管的搭建:將上述沉積物放入電爐中加熱至200-240℃保溫2-3小時,使沉積物中石墨稀與氧化鋅微米棒界面分離,然後將沉積物浸入質量濃度為15-20%的氫氧化鈉溶液中40-60分鐘,將沉積物中的ZnO微米棒溶解去除;過濾後用去離子水洗滌,在乾燥箱中40-60℃下烘乾9-12小時。
優選的,步驟b1中所述沉降液中網狀聯接石墨烯的濃度為150g/L-170g/L、氧化鋅微米棒的濃度為240g/L-280g/L、MoS2納米顆粒的濃度為90g/L-130g/L、葡萄糖的濃度為1.5g/L-4g/L、鉬酸鈉的濃度為10g/L-20g/L、磷酸二氫銨的濃度為40g/L-50g/L。
本發明的積極效果:根據本發明方法製備的孔隙型微米管結構催化儲氫協同功效材料,其由表面改性石墨烯形成的網絡聯接構建的微米管結構顯著地提高了石墨烯和MoS2納米顆粒的表面積,增大了氫氣的吸附區域。這種微米管結構材料具有中空結構,微米管壁具有連通孔隙,利於氫氣在微米管中的流動。MoS2納米顆粒固定於微米管壁石墨烯網絡內,可促使水分子在MoS2空位處分解成氧和氫原子,氫原子即被石墨烯吸附。因而本發明方法製備的孔隙型微米管結構材料既具有對電解水製備氫氣的催化效應,也具有高效吸附儲氫功能。
附圖說明
圖1是本發明所述催化儲氫協同功效材料製備方法的流程示意圖;
圖2是本發明所述催化儲氫協同功效材料的結構示意圖。
具體實施方式
下面結合附圖對本發明的優選實施例進行詳細說明。
參照圖1和圖2,本發明優選實施例提供一種孔隙型微米管結構催化儲氫協同功效材料的製備方法,按下列步驟順序進行:
①石墨烯表面羧基基團改性:將石墨烯、分析純的正已酸酐、鄰苯二甲酸酐加入乙烯乙二醇醚中,形成石墨烯濃度為60g/L-110g/L、正已酸酐濃度為160mL/L-200mLg/L和鄰苯二甲酸酐濃度為40g/L-60g/L的羧基改性液,超聲波攪拌20-40分鐘,在溫度為30-70℃環境下恆溫反應0.5-1.5小時,冷卻至室溫,過濾固相物質,用丙酮洗滌,在乾燥箱中40-60℃下乾燥8-12小時。
②石墨烯表面羥基基團改性:將石墨烯、分析純的磷酸二氫鉀、氫氧化鋇和乙酸亞鐵加入去離子水獲得羥基改性液,羥基改性液中的石墨烯濃度為1000g/L-130g/L、磷酸二氫鉀濃度為20g/L-65g/L、氫氧化鋇濃度為10g/L-20g/L、乙酸亞鐵濃度為30g/L-45g/L,超聲波攪拌30-40分鐘,在室溫反應1-1.5小時,過濾固相物質,用去離子水洗滌,在乾燥箱中50-60℃下乾燥10-13小時。
③石墨烯網狀聯接:將表面羧基基團和羥基基團改性的石墨烯各100-120g、分析純的三苯基磷-多滷代甲烷20-40g和二環已基碳二亞胺5-10g加入600-800mL的乙二醇單甲醚中,超聲波攪拌5-7分鐘,在室溫反應20-35分鐘,過濾固相物質,用丙酮洗滌後再以去離子水清洗,在乾燥箱中40-60℃下乾燥16-18小時,獲得網狀連接石墨烯。
④MoS2納米顆粒在空間網狀石墨烯沉降固定:將網狀聯接石墨烯、MoS2納米顆粒、氧化鋅微米棒、分析純的葡萄糖、鉬酸鈉和磷酸二氫銨加入去離子水形成網狀聯接石墨烯濃度為150g/L-170g/L、氧化鋅微米棒濃度為240g/L-280g/L、MoS2納米顆粒濃度為90g/L-130g/L、葡萄糖濃度為1.5g/L-4g/L、鉬酸鈉濃度為10g/L-20g/L和磷酸二氫銨濃度為40g/L-50g/L的沉降液;超聲波攪拌1-1.5小時,待沉降液固體沉澱後,過濾得沉積物。
⑤孔隙型微米管的搭建:將上述沉積物放入電爐中加熱至200-240℃保溫2-3小時,使沉積物中石墨稀與氧化鋅微米棒界面分離,將該沉積物浸入質量濃度為15-20%的氫氧化鈉溶液中40-60分鐘,將沉積物中的ZnO微米棒溶解去除;過濾後用去離子水洗滌,在乾燥箱中40-60℃下烘乾9-12小時。
下面給出具體實施例:
實施例:
①石墨烯表面羧基基團改性:將石墨烯、分析純的正已酸酐、鄰苯二甲酸酐加入乙烯乙二醇醚,形成石墨烯濃度為75g/L、正已酸酐濃度為160mL/L和鄰苯二甲酸酐濃度為50g/L的羧基改性液,超聲波攪拌40分鐘,在溫度為50℃環境下恆溫反應1小時,冷卻至室溫,過濾固相物質,用丙酮洗滌,在乾燥箱中40℃下乾燥9小時。
②石墨烯表面羥基基團改性:將石墨烯、分析純的磷酸二氫鉀、氫氧化鋇和乙酸亞鐵加入去離子水,使羥基改性液中的石墨烯濃度為110g/L、磷酸二氫鉀濃度為60g/L、氫氧化鋇濃度為10g/L和乙酸亞鐵濃度為35g/L,超聲波攪拌40分鐘,在室溫反應1小時,過濾固相物質,用去離子水洗滌,在乾燥箱中50℃下乾燥10小時。
③石墨烯網狀聯接:將表面羧基基團和羥基基團改性的石墨烯各120g、分析純的三苯基磷-多滷代甲烷30g和二環已基碳二亞胺10g加入700mL的乙二醇單甲醚中,超聲波攪拌7分鐘,在室溫反應25分鐘,過濾固相物質,用丙酮洗滌後再以去離子水清洗,在乾燥箱中60℃下乾燥16小時,獲得網狀連接石墨烯。
④MoS2納米顆粒在空間網狀石墨烯沉降固定:將網狀聯接石墨烯、MoS2納米顆粒、氧化鋅微米棒、分析純的葡萄糖、鉬酸鈉和磷酸二氫銨加入去離子水形成網狀聯接石墨烯濃度為160g/L、氧化鋅微米棒濃度為240g/L、MoS2納米顆粒濃度為110g/L、葡萄糖濃度為4g/L、鉬酸鈉濃度為12g/L和磷酸二氫銨濃度為45g/L的沉降液;超聲波攪拌1小時,待沉降液固體沉澱後,過濾得沉積物。
⑤孔隙型微米管的搭建:將上述沉積物放入電爐中加熱至220℃保溫2小時,使沉積物中石墨稀與氧化鋅微米棒界面分離,將該沉積物浸入質量濃度為20%的氫氧化鈉溶液中50分鐘,將沉積物中的ZnO微米棒溶解去除;過濾後用去離子水洗滌,在乾燥箱中60℃下烘乾9小時。
將本實施例製備的孔隙型微米管結構催化儲氫協同功效材料在常壓下儲氫時,其氫氣吸附容量達到7.1%,明顯高於儲氫合金LaNi5的2.1%。在電解水實驗中,孔隙型微米管結構材料作為陰極的交換電流密度比常用的鎳鉬合金大三倍。顯然,依據本發明製備的孔隙型微米管結構催化儲氫協同功效材料既具有對電解水製備氫氣的催化效應,也具有高效吸附儲氫功能。
以上所述的僅為本發明的優選實施例,所應理解的是,以上實施例的說明只是用於幫助理解本發明的方法及其核心思想,並不用於限定本發明的保護範圍,凡在本發明的思想和原則之內所做的任何修改、等同替換等等,均應包含在本發明的保護範圍之內。