一種3‑氨基‑5‑甲硫基‑1,2,4‑三氮唑的製備方法與流程
2023-07-19 04:03:11 4
本發明屬於抗病毒藥物化學合成技術領域,特別涉及一種3-氨基-5-甲硫基-1,2,4-三氮唑的製備方法。
背景技術:
3-氨基-5-甲硫基-1,2,4-三氮唑是一種新型抗病毒藥物的重要原料,可用於合成具有廣譜抗病毒活性的三唑-三嗪類藥物。這類藥物能有效治療克裡米亞-剛果出血熱、裂谷熱、西尼羅河病毒感染等疾病。可以保護動物(60%-90%)免受流感病毒、蜱傳播的腦炎、呼吸道感染以及其它多種社會影響大、危害高的病毒性傳染病。在抑制高致病人類流感病毒的再生方面療效顯著,包括H1N1、H3N5、H5N1和非流感病毒如西部馬腦脊髓炎病毒、呼吸道合胞病毒、雞傳染性喉氣管炎病毒、禽流感病毒等,能夠用於預防和治療人類和動物病毒感染性疾病。該化合物具有抗病毒活性高,作用持續時間長和毒性低等特點。
到目前為止,僅有CN201310116564-一種5-氨基-3-巰基-1,2,4-三氮唑晶體及其製備方法公開了一種利用5-氨基-3-巰基-1,2,4三氮唑和碘甲烷為原料,進行甲基化反應得到產品的製備方法,儘管該方法相對其他現有製備方法已經具有一定環境汙染減少,成本降低,純度提高等效果,但其仍然存在諸多弊端。
目前的3-氨基-5-甲硫基-1,2,4-三氮唑多是以5-氨基-3-巰基-1,2,4三氮唑和碘甲烷為原料,進行甲基化反應得到產品,次工藝所用原料比較昂貴,成本高,且會產生大量含碘廢水,很難處理,工業化放大受到限制。
三唑雜環化合物具有廣泛的生物活性,如抗菌,鎮靜,抗焦慮等作用,因此,一直是雜環化合物的中藥研究領域,其中3-氨基-5-甲硫基-1,2,4-三氮唑是合成三唑雜環衍生物的重要中間體。
因此提供一種新的3-氨基-5-甲硫基-1,2,4-三氮唑的合成方法,節省成本的同時減少環境汙染,便於工業化大範圍加工就顯得尤為必要。
技術實現要素:
為解決上述現有技術存在的問題,本發明的目的在於提供一種3-氨基-5-甲硫基-1,2,4-三氮唑的製備方法,以氰氨基二硫化碳酸二甲酯、水合肼為原料,以醇或水作為溶劑,通過一步反應製備合成目標產物,原料成本低,不產生汙染環境的含碘廢水,操作步驟簡單,且產物純度能達到98%以上,具有很好的商業應用前景,值得大範圍推廣。
為達到上述目的,本發明的技術方案為:
一種3-氨基-5-甲硫基-1,2,4-三氮唑的製備方法,反應器進行氮氣置換後,以氰氨基二硫化碳酸二甲酯、水合肼為原料,溶劑為醇類有機溶劑,將氰氨基二硫化碳酸二甲酯加入溶劑中攪拌溶解後,在25-28℃條件下,緩慢滴加水合肼,氰氨基二硫化碳酸二甲酯和水合肼摩爾比為1:1.1-1:15,滴加1.5-2小時,之後升溫至65-70℃,在回流條件下,反應2-3小時,通過一步反應製備合成類白色固體,經核磁確認為目標產物3-氨基-5-甲硫基-1,2,4-三氮唑,反應方程式為:
進一步的,所述有機溶劑為甲醇或乙醇。
進一步的,所述方法3-氨基-5-甲硫基-1,2,4-三氮唑的純度≥98%。
進一步的,所述溶劑的體積為氰氨基二硫化碳酸二甲酯重量的5-7倍。
進一步的,所述方法反應合成收率≥90%。
相對於現有技術,本發明的有益效果為:
本發明一種3-氨基-5-甲硫基-1,2,4-三氮唑的製備方法,以氰氨基二硫化碳酸二甲酯、水合肼為原料,以醇或水作為溶劑,通過一步反應製備合成目標產物,原料成本低,不產生汙染環境的含碘廢水,操作步驟簡單,且產物純度能達到98%以上,具有很好的商業應用前景,值得大範圍推廣。
附圖說明
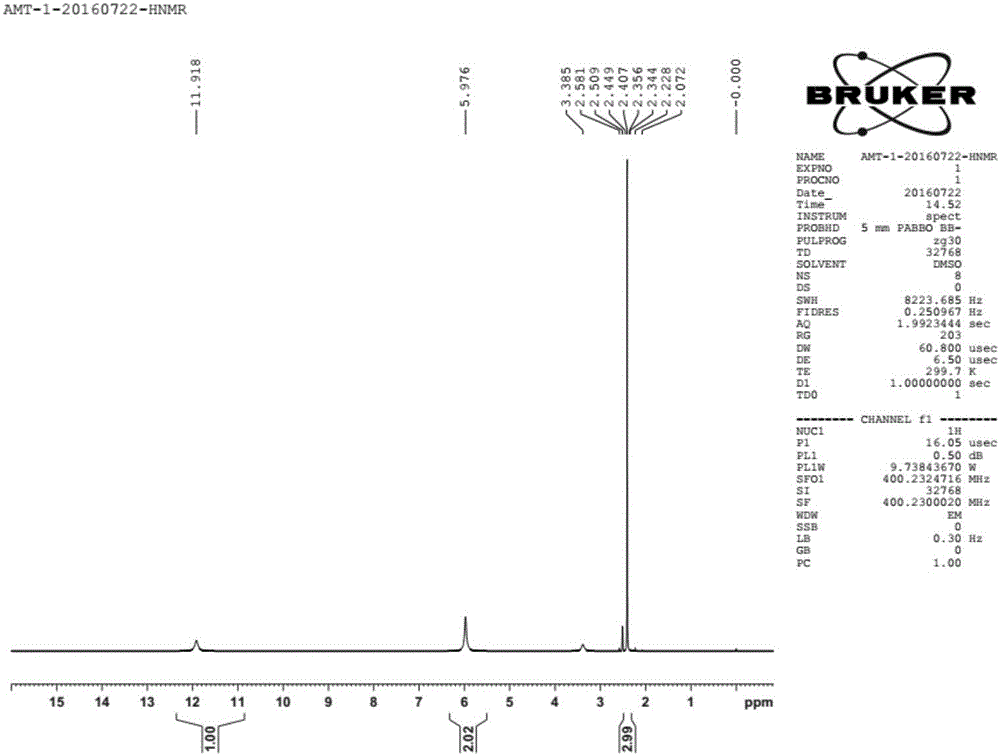
圖1為目標產物的氫核磁共振HNMR圖譜。
圖2為目標產物的碳核磁共振CNMR圖譜。
圖3為目標產物的液質聯用LC-MS圖譜。
具體實施方式
下面結合實施例對本發明做進一步的說明,以下所述,僅是對本發明的較佳實施例而已,並非對本發明做其他形式的限制,任何熟悉本專業的技術人員可能利用上述揭示的技術內容加以變更為同等變化的等效實施例。凡是未脫離本發明方案內容,依據本發明的技術實質對以下實施例所做的任何簡單修改或等同變化,均落在本發明的保護範圍內。
如圖1-3所示,一種3-氨基-5-甲硫基-1,2,4-三氮唑的製備方法,反應器進行氮氣置換後,以氰氨基二硫化碳酸二甲酯、水合肼為原料,溶劑為醇類有機溶劑,將氰氨基二硫化碳酸二甲酯加入溶劑中攪拌溶解後,在25-28℃條件下,緩慢滴加水合肼,氰氨基二硫化碳酸二甲酯和水合肼摩爾比為1:1.1-1:15,滴加1.5-2小時,之後升溫至65-70℃,在回流條件下,反應2-3小時,通過一步反應製備合成類白色固體,經核磁確認為目標產物3-氨基-5-甲硫基-1,2,4-三氮唑,反應方程式為:
進一步的,所述有機溶劑為甲醇或乙醇。
進一步的,所述方法3-氨基-5-甲硫基-1,2,4-三氮唑的純度≥98%。
進一步的,所述溶劑的體積為氰氨基二硫化碳酸二甲酯重量的5-7倍。
進一步的,所述方法反應合成收率≥90%。
以下結合數個較佳實施例對本發明技術方案作進一步非限制性的詳細說明。
實施例1
反應器進行氮氣置換後,加入250ml甲醇,50g氰氨基二硫化碳酸二甲酯,攪拌溶解後,自然升至室溫,在25℃攪拌下,然後緩慢滴加32g水合肼,滴加時間保持在1.5小時,然後升溫至65℃至回流反應2小時,然後反應液降溫至10℃,過濾,濾餅用水洗滌,乾燥後得到類白色固體40g,反應方程式為:
,液相含量為99.2%。
實施例2
反應器進行氮氣置換後,加入1323ml乙醇,189g氰氨基二硫化碳酸二甲酯,攪拌溶解後,自然升至室溫,在28℃攪拌下,然後緩慢滴加96g水合肼,滴加時間保持在1.8小時,然後升溫至67℃至回流2.5小時,冷卻至10℃,過濾,濾餅用水洗滌,乾燥後得到類白色固體134g,反應方程式為:
,液相含量為99.5%,所得產物的氫核磁共振HNMR圖譜如圖1所示。碳核磁共振CNMR圖譜如圖2所示。液質聯用LC-MS圖譜如圖3所示。
實施例3
反應器進行氮氣置換後,加入350ml甲醇,50g氰氨基二硫化碳酸二甲酯,攪拌溶解後,自然升至室溫,在27℃攪拌下,然後緩慢滴加40g水合肼,滴加時間保持在2小時,然後升溫至70℃至回流3小時,冷卻至10℃,過濾,濾餅用水洗滌,乾燥後得到類白色固體32g,反應方程式為:
,液相含量為98.5%。
以上所述,僅為本發明的具體實施方式,但本發明的保護範圍並不局限於此,任何不經過創造性勞動想到的變化或替換,都應涵蓋在本發明的保護範圍之內。因此,本發明的保護範圍應該以權利要求書所限定的保護範圍為準。