一種複合阻燃劑的製備方法與流程
2023-12-03 17:12:22 2
本發明涉及高分子材料的無滷阻燃技術領域,特別涉及一種複合阻燃劑的製備方法。
背景技術:
9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO)是一種重要的無滷含磷阻燃劑,其分子具有菲環結構,分子中的P-C鍵水解穩定性好,P-O鍵鍵能大,熱穩定性高,聯苯結構具有較高的剛性。相比滷系阻燃劑,DOPO及其衍生物在燃燒過程中所釋放出的腐蝕性和毒性氣體很少,是一類性能優良、用途廣泛的無滷阻燃劑。
石墨烯是由單層碳原子以六角形sp2雜化方式形成的二維結構碳材料,具有超乎其它材料的物理化學性能,如熱傳導性能好、楊氏模量高、導電性能好,在製備電池、電磁屏蔽、納(微)米器件、清潔能源等領域展現出極其廣泛的應用前景。同時,石墨烯的層狀結構可以阻止可燃氣體逸出到火焰區、阻止能量回傳且促進成炭,有望成為高效低添加量的新型阻燃劑。
將DOPO與石墨烯或氧化石墨烯進行接枝,得到DOPO改性石墨烯或DOPO改性氧化石墨烯,與一般石墨烯或氧化石墨烯相比在有機溶劑中具有更好的分散穩定性,作為阻燃劑應用於環氧樹脂、聚碳酸醋等聚合物基體中時,能夠顯著提高聚合物材料的阻燃性能。現有技術中將DOPO與石墨烯或氧化石墨烯進行接枝前,通常需要對石墨烯或氧化石墨烯進行改性處理,如將氧化石墨烯用氯化亞碸處理,使氧化石墨烯上的羧基被氯化形成醯氯基,利用DOPO中磷氫鍵(P-H)易斷開的特性,與醯氯基發生反應,使得DOPO被修飾到石墨烯上;又如用含異氰酸根的矽烷先對石墨烯進行改性,然後通過溶膠凝膠法再引入DOPO,製備得到含DOPO和層離石墨烯的納米複合阻燃劑。但是,上述方法操作複雜,實際應用過程中局限性較大,不利於大規模生產。
技術實現要素:
本發明的目的在於提供一種複合阻燃劑的製備方法,以期通過簡單方便的操作製備得到具有較好阻燃效果的複合阻燃劑。
本發明提供了一種複合阻燃劑的製備方法,所述複合阻燃劑為9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯,包括以下步驟:
(1)提供氧化石墨烯的四氫呋喃分散液體系和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物的四氫呋喃溶液體系;
(2)將所述步驟(1)中的分散液體系和溶液體系混合,在保護氣氛下進行接枝反應,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯。
優選的,所述步驟(1)中的分散液體系在超聲條件下得到。
優選的,所述超聲的時間為30~60min,功率為80~100W,頻率為80~100kHz。
優選的,所述步驟(1)中的分散液體系中氧化石墨烯的質量濃度為2.5~10g/L。
優選的,所述步驟(1)中的溶液體系中9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液的質量濃度為13~60g/L。
優選的,所述步驟(2)中混合得到的混合物料中,氧化石墨烯的質量、9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物的質量與四氫呋喃的體積比為(1~2)g:(4~6)g:(0.3~0.7)L。
優選的,步驟(2)中所述接枝反應的溫度為65~70℃,時間為10~12h。
優選的,步驟(2)中所述接枝反應在攪拌條件下進行,所述攪拌的速率為60~100rpm。
優選的,步驟(2)所述接枝反應完成後還包括:對接枝反應後得到的物料依次進行洗滌、離心和乾燥,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯。
優選的,所述乾燥的溫度為80~100℃,時間為10~15h。
本發明提供了一種複合阻燃劑的製備方法,首先提供氧化石墨烯的四氫呋喃分散液體系和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物的四氫呋喃溶液體系;然後將分散液體系和溶液體系混合,在保護氣氛下進行接枝反應,得到複合阻燃劑。本發明提供的製備方法操作簡單方便,有利於實現大規模生產。採用本發明提供的製備方法得到的複合阻燃劑具有較好的阻燃效果,為未來協效阻燃提供了更多的選擇,拓展了石墨烯在改善高分子材料阻燃性能方面的應用。實驗結果表明,將本發明提供的複合阻燃劑應用於高分子材料中,能夠使得到的複合材料的阻燃級別達到UL-94V0級,氧指數能夠達到24.2%,明顯高於純EP的氧指數。
附圖說明
下面結合附圖和具體實施方式對本發明作進一步詳細的說明。
圖1為9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物與實施例1製備的9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯和氧化石墨烯的傅立葉變換紅外光譜圖(FT-IR譜圖);
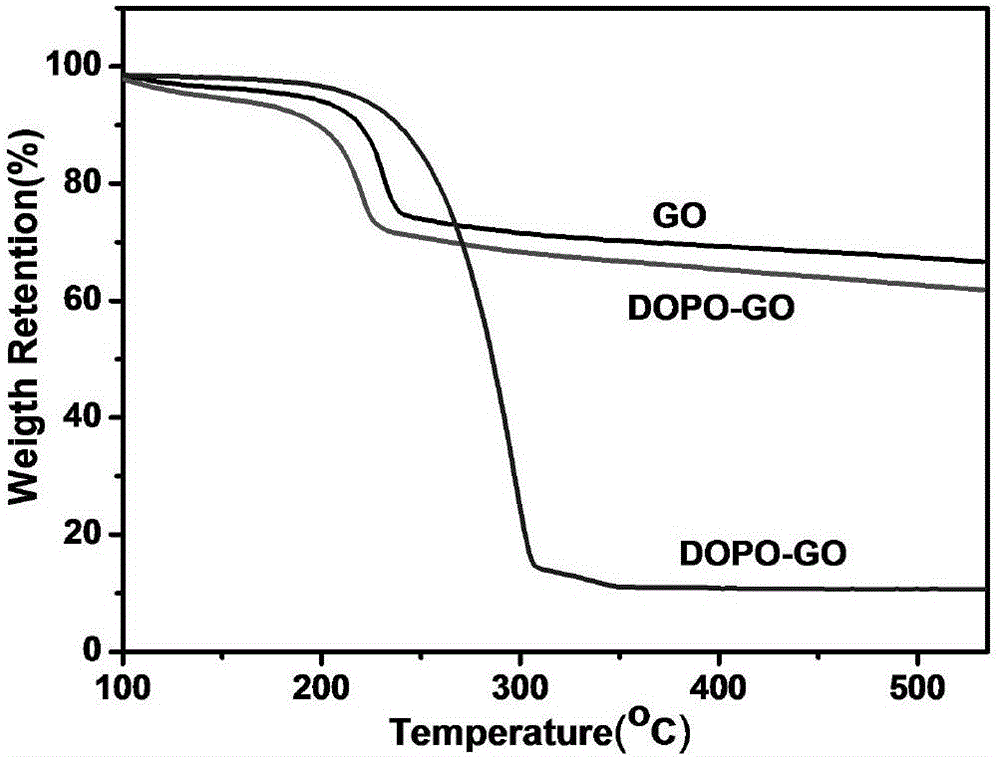
圖2為9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物與實施例1製備的9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯和氧化石墨烯的熱重分析曲線(TGA曲線);
圖3為9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物與實施例1製備的9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯和氧化石墨烯的X射線衍射圖(XRD圖)。
具體實施方式
本發明提供了一種複合阻燃劑的製備方法,所述複合阻燃劑為9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯,包括以下步驟:
(1)提供氧化石墨烯的四氫呋喃分散液體系和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物的四氫呋喃溶液體系;
(2)將所述步驟(1)中的分散液體系和溶液體系混合,在保護氣氛下進行接枝反應,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯。
本發明提供氧化石墨烯的四氫呋喃分散液體系。在本發明中,所述分散液體系中氧化石墨烯的質量濃度優選為2.5~10g/L,更優選為4~8g/L,最優選為5~7g/L。本發明對於所述分散液體系的製備方法沒有特殊的限定,採用本領域技術人員熟知的製備分散液體系的技術方案即可。在本發明中,所述分散液體系優選在超聲條件下得到,具體是將氧化石墨烯與四氫呋喃混合,超聲分散,得到所述分散液體系。在本發明中,所述超聲的時間優選為30~60min,更優選為35~55min,最優選為40~50min;所述超聲的功率優選為80~100W,更優選為85~95W,最優選為88~92W;所述超聲的頻率優選為80~100kHz,更優選為85~95kHz,最優選為88~92kHz。
本發明對於所述氧化石墨烯的來源沒有特殊的限定,採用本領域技術人員熟知的市售商品或者是能夠製備得到氧化石墨烯的技術方案即可。本發明優選採用改進的預氧化-Hummers法製備氧化石墨烯(GO),包括以下步驟:
(1)將石墨與濃硫酸、K2S2O8和P2O5混合,在60~80℃條件下進行預氧化反應,得到預氧化石墨;
(2)將所述步驟(1)得到的預氧化石墨與濃硫酸和KMnO4混合,在35~40℃條件下進行氧化反應;
(3)將所述步驟(2)得到的物料與水混合,在90~95℃下進行反應,然後加入雙氧水繼續進行反應,得到氧化石墨烯。
本發明將石墨與濃硫酸、K2S2O8和P2O5混合,在60~80℃條件下進行預氧化反應,得到預氧化石墨。在本發明中,所述石墨與濃硫酸、K2S2O8和P2O5的質量比優選為1:(0.9~1.1):(0.4~0.6):(0.4~0.6),更優選為1:1:0.5:0.5。在本發明中,所述石墨優選為石墨粉;所述石墨粉的粒度優選為325~500目,更優選為400~460目。在本發明中,所述濃硫酸的質量濃度優選為95%~98%。本發明優選將濃硫酸、K2S2O8和P2O5混合後,再與石墨混合。在本發明中,所述混合優選在攪拌條件下進行,所述攪拌的速率優選為60~100rpm,更優選為70~85rpm。在本發明中,所述預氧化反應的溫度為60~80℃,優選為65~75℃,更優選為62~72℃;所述預氧化反應的時間優選為4~6h,更優選為4.5~5.5h。在本發明中,所述預氧化反應優選在油浴條件下進行。
完成所述預氧化反應後,本發明優選將預氧化反應產物進行後處理,得到預氧化石墨。在本發明中,所述後處理優選包括以下步驟:
將預氧化反應產物冷卻、洗滌、抽濾、乾燥,得到預氧化石墨。
本發明對於所述冷卻沒有特殊的限定,採用本領域技術人員熟知的冷卻的技術方案即可。在本發明的實施例中,具體是將預氧化反應產物自然冷卻至室溫。本發明對於所述洗滌沒有特殊的限定,採用本領域技術人員熟知的洗滌的技術方案即可。在本發明的實施例中,具體是採用去離子水將冷卻後的預氧化反應產物洗滌至中性。本發明對於所述抽濾沒有特殊的限定,採用本領域技術人員熟知的能夠實現固液分離的抽濾的技術方案即可。本發明對於所述乾燥沒有特殊的限定,採用本領域技術人員熟知的乾燥的技術方案即可。在本發明中,所述乾燥的時間優選為15~20h,更優選為16~18h;所述乾燥的溫度優選為20~40℃,更優選為25~35℃。在本發明的實施例中,具體是將洗滌後的預氧化反應產物於室溫下進行自然乾燥。
得到預氧化石墨後,本發明將所述預氧化石墨與濃硫酸和KMnO4混合,在35~40℃條件下進行氧化反應。在本發明中,所述預氧化石墨、濃硫酸和KMnO4的質量比優選為1:(50~60):(2.5~3.5),更優選為1:(54~56):3。在本發明中,所述濃硫酸的質量濃度優選為95%~98%。本發明優選將濃硫酸和KMnO4混合後,再與預氧化石墨混合。在本發明中,所述混合優選在攪拌條件下進行,所述攪拌的速率優選為60~100rpm,更優選為70~85rpm。本發明優選在4~6℃條件下將濃硫酸和KMnO4混合。在本發明中,所述氧化反應的溫度為35~40℃,優選為36~38℃;所述氧化反應的時間優選為2~3h,更優選為2.5h。在本發明中,所述氧化反應優選在油浴條件下進行。
完成所述氧化反應後,本發明將氧化反應後得到的物料與水混合,在90~95℃下進行反應,然後加入雙氧水繼續進行反應,得到氧化石墨烯。在本發明中,所述水與氧化反應步驟中所加入的濃硫酸的質量比優選為(1.5~2.5):1,更優選為2:1。本發明對於所述水沒有特殊的限定,採用本領域技術人員熟知的水即可;在本發明的實施例中,具體採用去離子水。在本發明中,所述混合優選在攪拌條件下進行,所述攪拌的速率優選為60~100rpm,更優選為70~85rpm。在本發明中,將氧化反應後得到的物料與水混合後進行反應的溫度為90~95℃,優選為92~94℃;所述反應的時間優選為30~45min,更優選為35~40min。在本發明中,所述反應優選在油浴條件下進行。
完成所述氧化反應後得到的物料與水的反應後,本發明加入雙氧水繼續進行反應,得到氧化石墨烯。在本發明中,所述雙氧水的質量濃度優選為28~30%。在本發明中,所述雙氧水用於除去反應液中多餘的KMnO4;本發明對於所述雙氧水的加入量沒有特殊的限定,根據實際情況,採用本領域技術人員熟知的能夠使最終反應液呈金黃色的雙氧水的加入量即可。
加入雙氧水繼續進行反應完成後,本發明優選將反應得到的物料進行後處理,得到氧化石墨烯。在本發明中,所述後處理優選包括以下步驟:
將反應得到的物料進行洗滌、抽濾、乾燥,得到氧化石墨烯。
本發明對於所述洗滌沒有特殊的限定,採用本領域技術人員熟知的洗滌的技術方案即可。本發明優選採用鹽酸洗滌反應得到的物料,然後用去離子水繼續洗滌至中性。在本發明中,所述鹽酸的質量濃度優選為8~12%。本發明對於所述抽濾沒有特殊的限定,採用本領域技術人員熟知的能夠實現固液分離的抽濾的技術方案即可。本發明對於所述乾燥沒有特殊的限定,採用本領域技術人員熟知的乾燥的技術方案即可。在本發明中,所述乾燥優選為冷凍乾燥;在本發明的實施例中,具體採用冷凍乾燥機進行所述乾燥。在本發明中,所述乾燥的溫度優選為0~5℃,更優選為2~4℃;所述乾燥的時間優選為2~10h,更優選為5~7h。
本發明提供9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物的四氫呋喃溶液體系。在本發明中,所述溶液體系中9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液的質量濃度優選為13~60g/L,更優選為22~48g/L,最優選為30~35g/L。本發明對於所述溶液體系的製備方法沒有特殊的限定,採用本領域技術人員熟知的製備溶液體系的技術方案即可。在本發明中,具體是將9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO)與四氫呋喃混合,得到所述溶液體系。
得到分散液體系和溶液體系後,本發明將所述分散液體系和溶液體系混合,在保護氣氛下進行接枝反應,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯(DOPO-GO)。在本發明中,所述混合得到的混合物料中,氧化石墨烯的質量、9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物的質量與四氫呋喃的體積比優選為(1~2)g:(4~6)g:(0.3~0.7)L,更優選為(1.3~1.7)g:(4.5~5.5)g:(0.35~0.65)L,再優選為1.5g:5g:(0.4~0.6)L,最優選為1.5g:5g:(0.45~0.55)L。
本發明對於提供所述保護氣氛的保護氣體的種類沒有特殊的限定,採用本領域技術人員熟知的保護氣體即可。在本發明的實施例中,具體選用氮氣作為保護氣體。在本發明中,所述接枝反應的溫度優選為65~70℃,所述接枝反應的時間優選為10~12h。在本發明中,所述接枝反應優選在攪拌條件下進行;所述攪拌的速率優選為60~100rpm,更優選為70~85rpm。
所述接枝反應完成後,本發明優選對接枝反應後得到的物料進行後處理,得到所述9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯。在本發明中,所述後處理優選包括以下步驟:
對接枝反應後得到的物料依次進行洗滌、離心和乾燥,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯。
本發明對於所述洗滌沒有特殊的限定,採用本領域技術人員熟知的洗滌的技術方案即可。本發明優選依次採用四氫呋喃和丙酮對反應後得到的物料洗滌。本發明對於所述離心沒有特殊的限定,採用本領域技術人員熟知的離心的技術方案即可。在本發明中,所述離心的轉速優選為8000~10000rpm,更優選為8500~9500rpm;所述離心的時間優選為1~2h,更優選為1.5h。本發明對於所述乾燥沒有特殊的限定,採用本領域技術人員熟知的乾燥的技術方案即可。在本發明中,所述乾燥的溫度優選為80~100℃,更優選為85~95℃;所述乾燥的時間優選為10~15h,更優選為11~13h。在本發明中,所述乾燥優選為真空乾燥。所述真空乾燥的真空度優選為0.09~0.1MPa。
下面將結合本發明中的實施例,對本發明中的技術方案進行清楚、完整地描述。顯然,所描述的實施例僅僅是本發明一部分實施例,而不是全部的實施例。基於本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬於本發明保護的範圍。
實施例1
(1)將3mL質量濃度為98%的濃硫酸、3g K2S2O8和3g P2O5在75rpm條件下攪拌混合,然後加入5g石墨粉,油浴升溫至70℃,繼續攪拌進行5h預氧化反應,然後將預氧化反應產物自然冷卻至室溫,採用去離子水將冷卻後的預氧化反應產物洗滌至中性,抽濾,將得到的固體於室溫下自然乾燥17h,得到預氧化石墨;
(2)將146mL質量濃度為98%的濃硫酸和15g KMnO4在5℃、75rpm條件下攪拌混合,加入5g所述步驟(1)得到的預氧化石墨,攪拌混合3h,油浴升溫至36℃,繼續攪拌進行2.5h氧化反應;
(3)向所述步驟(2)得到的物料中加入250mL去離子水,油浴升溫至93℃,在75rpm條件下攪拌反應35min,然後加入40mL質量濃度為30%的雙氧水繼續進行反應,至反應液呈金黃色;用100mL質量濃度為10%的鹽酸洗滌反應得到的物料,然後用去離子水繼續洗滌至中性,抽濾,採用冷凍乾燥機將得到的固體於3℃下冷凍乾燥7h,得到氧化石墨烯;
(4)將0.6g所述步驟(3)得到的氧化石墨烯和200mL四氫呋喃混合,在功率為90W、頻率為90kHz的條件下超聲45min,得到氧化石墨烯分散液;將2g 9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物與100mL四氫呋喃混合,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液;
(5)將步驟(4)得到的氧化石墨烯分散液和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液混合,油浴升溫至70℃,氮氣保護、75rpm條件下攪拌反應12h;依次用四氫呋喃和丙酮洗滌反應後得到的物料,9000rpm下離心1.5h,在真空度為0.09MPa、90℃條件下真空乾燥12h,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯(DOPO-GO)。
對本發明實施例1製備得到的9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯(DOPO-GO)和氧化石墨烯(GO)進行傅立葉變換紅外光譜(FT-IR)分析,並與9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物(DOPO)進行比較,結果見圖1。由圖1可知,DOPO的主要特徵峰是2300cm-1(P-H)、1446cm-1(P-Ph)、1210cm-1(P=O)和755cm-1(P-O-Ph);GO的主要特徵峰是C=O、C=C、環氧環和羧基等含氧基團。將GO與DOPO進行接枝實際上是使DOPO分子中的P-H與GO表面上的環氧環打開,使P與GO表面上環氧環的C形成共價鍵,H與O共價結合形成羥基(-OH),從而將DOPO分子接枝在GO的表面。但是由於分子是通過共價鍵結合,所以在DOPO-GO中仍會同時存在一些GO或DOPO的特徵峰。圖1中DOPO-GO的FT-IR譜圖包括P-Ph、P=O、C=O、C=C等特徵峰,而P-H與環氧環的特徵峰相應減弱或者是完全消失,這進一步說明DOPO接枝在GO的表面是通過斷裂P-H與環氧環實現的,也說明了本發明實施例1製備的DOPO-GO將DOPO接枝到了GO的表面上。
對DOPO與實施例1製備的DOPO-GO和GO進行熱重分析,結果見圖2。由圖2可知,GO的起始分解溫度約為175℃,這是由於GO表面含有許多含氧基團,如-OH、-COOH、-CO等,隨著溫度的升高,這些基團開始分解,生成難燃的CO2、CO和H2O;在200~300℃時分解的速率最快。DOPO-GO的起始分解溫度約為130℃,較GO降低了45℃;到175℃時分解速率加快,在190~250℃時分解的速率最快,最大的失重溫度為217℃。這是由於在GO表面接枝了DOPO後,隨著溫度的升高,DOPO-GO表面開始碳化,並開始進行熱與能量的交換,當達到一定溫度值時,釋放出氣體,如水蒸氣,對整個燃燒體系起到了冷凝作用;另外,水蒸氣還稀釋了體系中的可燃氣體和氧氣的濃度,同時DOPO-GO會生成PO·,可以捕獲高分子材料燃燒後釋放出的活性自由基H·和OH·,達到中斷鏈式反應的效果。通過數據分析,DOPO在530℃時的殘炭量僅為10%,而DOPO-GO在530℃時的殘炭量約為60%,增加了50%,說明碳層在燃燒過程中大大阻隔了可燃氣體的釋放和熱量的交換,起到了阻燃的目的。
對DOPO與實施例1製備的DOPO-GO和GO進行X射線衍射分析,結果見圖3。由圖3可知,GO對應的特徵峰為11.3°,層間距為0.78nm;DOPO-GO對應的特徵峰為10.7°,層間距為0.83nm。說明功能化的官能團存在與DOPO-GO片層表面。
將實施例1製備的DOPO-GO添加到環氧樹脂(EP)中,製備得到DOPO-GO/EP複合材料,按照GB/T 2408-2008規定的方法測試阻燃級別,按照GB/T 2406.1-2008規定的方法測試氧指數,並與純環氧樹脂的阻燃性能進行比較,結果見表1;其中,按佔所述環氧樹脂的質量百分比計,加入1%DOPO-GO得到的複合材料計為1%DOPO-GO/EP,加入2.5%DOPO-GO得到的複合材料計為2.5%DOPO-GO/EP。
表1DOPO-GO/EP複合材料與純環氧樹脂的阻燃性能數據
由表1可以看出,實施例1提供的複合阻燃劑DOPO-GO具有較好的阻燃效果,應用於高分子材料中,能夠使得到的複合材料的阻燃級別達到UL-94V0級,氧指數能夠達到24.2%,明顯高於純EP的氧指數。
實施例2
(1)將3.5mL質量濃度為98%的濃硫酸、3g K2S2O8和3g P2O5在100rpm條件下攪拌混合,然後加入6g石墨粉,油浴升溫至80℃,繼續攪拌進行4h預氧化反應,然後將預氧化反應產物自然冷卻至室溫,採用去離子水將冷卻後的預氧化反應產物洗滌至中性,抽濾,將得到的固體於室溫下自然乾燥15h,得到預氧化石墨;
(2)將180mL質量濃度為98%的濃硫酸和18g KMnO4在5℃、100rpm條件下攪拌混合,加入6g所述步驟(1)得到的預氧化石墨,攪拌混合4h,油浴升溫至35℃,繼續攪拌進行3h氧化反應;
(3)向所述步驟(2)得到的物料中加入250mL去離子水,油浴升溫至90℃,在100rpm條件下攪拌反應30min,然後加入50mL質量濃度為30%的雙氧水繼續進行反應,至反應液呈金黃色;用150mL質量濃度為10%的鹽酸洗滌反應得到的物料,然後用去離子水繼續洗滌至中性,抽濾,採用冷凍乾燥機將得到的固體於0℃下冷凍乾燥2h,得到氧化石墨烯;
(4)將0.9g所述步驟(3)得到的氧化石墨烯和200mL四氫呋喃混合,在功率為80W、頻率為100kHz的條件下超聲60min,得到氧化石墨烯分散液;將3g 9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物與150mL四氫呋喃混合,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液;
(5)將步驟(4)得到的氧化石墨烯分散液和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液混合,油浴升溫至70℃,氮氣保護、100rpm條件下攪拌反應12h;依次用四氫呋喃和丙酮洗滌反應後得到的物料,10000rpm下離心1h,在真空度為0.1MPa、80℃條件下真空乾燥12h,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯(DOPO-GO)。
實施例3
(1)將4mL質量濃度為98%的濃硫酸、3.5g K2S2O8和3.5g P2O5在60rpm條件下攪拌混合,然後加入7g石墨粉,油浴升溫至60℃,繼續攪拌進行6h預氧化反應,然後將預氧化反應產物自然冷卻至室溫,採用去離子水將冷卻後的預氧化反應產物洗滌至中性,抽濾,將得到的固體於室溫下自然乾燥20h,得到預氧化石墨;
(2)將190mL質量濃度為98%的濃硫酸和21g KMnO4在5℃、60rpm條件下攪拌混合,加入7g所述步驟(1)得到的預氧化石墨,攪拌混合3h,油浴升溫至40℃,繼續攪拌進行2h氧化反應;
(3)向所述步驟(2)得到的物料中加入250mL去離子水,油浴升溫至95℃,在60rpm條件下攪拌反應45min,然後加入40mL質量濃度為30%的雙氧水繼續進行反應,至反應液呈金黃色;用100mL質量濃度為10%的鹽酸洗滌反應得到的物料,然後用去離子水繼續洗滌至中性,抽濾,採用冷凍乾燥機將得到的固體於5℃下冷凍乾燥10h,得到氧化石墨烯;
(4)將0.75g所述步驟(3)得到的氧化石墨烯和200mL四氫呋喃混合,在功率為100W、頻率為80kHz的條件下超聲30min,得到氧化石墨烯分散液;將2.5g 9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物與100mL四氫呋喃混合,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液;
(5)將步驟(4)得到的氧化石墨烯分散液和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液混合,油浴升溫至65℃,氮氣保護、60rpm條件下攪拌反應10h;依次用四氫呋喃和丙酮洗滌反應後得到的物料,8000rpm下離心2h,在真空度為0.09MPa、100℃條件下真空乾燥10h,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯(DOPO-GO)。
實施例4
(1)將5mL質量濃度為98%的濃硫酸、4g K2S2O8和4g P2O5在85rpm條件下攪拌混合,然後加入8g石墨粉,油浴升溫至65℃,繼續攪拌進行5.5h預氧化反應,然後將預氧化反應產物自然冷卻至室溫,採用去離子水將冷卻後的預氧化反應產物洗滌至中性,抽濾,將得到的固體於室溫下自然乾燥16h,得到預氧化石墨;
(2)將240mL質量濃度為98%的濃硫酸和20g KMnO4在4℃、85rpm條件下攪拌混合,加入8g所述步驟(1)得到的預氧化石墨,攪拌混合3h,油浴升溫至38℃,繼續攪拌進行2h氧化反應;
(3)向所述步驟(2)得到的物料中加入250mL去離子水,油浴升溫至92℃,在85rpm條件下攪拌反應40min,然後加入15mL質量濃度為30%的雙氧水繼續進行反應,至反應液呈金黃色;用100mL質量濃度為10%的鹽酸洗滌反應得到的物料,然後用去離子水繼續洗滌至中性,抽濾,採用冷凍乾燥機將得到的固體於2℃下冷凍乾燥5h,得到氧化石墨烯;
(4)將1g所述步驟(3)得到的氧化石墨烯和200mL四氫呋喃混合,在功率為85W、頻率為95kHz的條件下超聲50min,得到氧化石墨烯分散液;將3.5g9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物與100mL四氫呋喃混合,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液;
(5)將步驟(4)得到的氧化石墨烯分散液和9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物溶液混合,油浴升溫至70℃,氮氣保護、85rpm條件下攪拌反應11h;依次用四氫呋喃和丙酮洗滌反應後得到的物料,8000rpm下離心2h,在真空度為0.09MPa、95℃條件下真空乾燥12h,得到9,10-二氫-9-氧雜-10-磷雜菲-10-氧化物接枝氧化石墨烯(DOPO-GO)。
以上所述僅是本發明的優選實施方式,應當指出,對於本技術領域的普通技術人員來說,在不脫離本發明原理的前提下,還可以做出若干改進和潤飾,這些改進和潤飾也應視為本發明的保護範圍。