小麥BSR‑Seq基因定位的方法與流程
2023-12-10 04:09:01 3
本發明涉及一種遺傳育種技術,尤其涉及一種小麥BSR-Seq(Bulked Segregant RNA-Seq,混池轉錄組測序)基因定位的方法。
背景技術:
小麥是人類主要口糧作物之一,在耕地減少、需求量不斷上升的背景下,其產量豐欠攸關糧食安全。培育高產優質抗逆小麥品種和遺傳改良小麥重要農藝性狀是小麥穩產增產的重要方法。提高小麥傳統育種方法的效率、不斷地創新和應用新的育種技術是必經之路。當前分子標記技術、標記輔助選擇育種和基因組選擇愈發得到關注,利用緊密連鎖的分子標記鑑定和篩選與表型相關的染色體區段或基因對於基因精細定位和克隆、標記輔助選擇育種、基因聚合育種、分子設計育種具有重要意義和應用價值。然而,與小麥重要農藝性狀緊密連鎖的分子標記的開發面臨諸多挑戰,主要由於小麥缺乏基因組序列信息且基因組複雜,讓基於PCR的分子標記開發較為困難,來源於基因的PCR標記和RFLP標記常擴增或雜交到小麥的部分同源染色體上,因此常被定位到部分同源染色體上。當前有多種方法用於解決部分問題。
第一種方法是利用小麥染色體片段缺失系和缺體-四體系。四百多套中國春染色體片段缺失系得到開發(Endo等,1996),每個系都有一個染色體片段被刪除,有大量EST序列錨定在了特定刪除區段內(Qi等,2004),這可以通過細胞遺傳學的方法進行檢測。另外,一系列染色體缺體-四體系也得到開發,每個系的一對染色體被其部分同源的染色體替換。理想的染色體缺體-四體系只在含有目的基因的染色體被替換,由此構建的作圖群體使多態性標記定位到期望的染色體上,避免了定位到部分同源染色體上。如Fairs等用染色體片段缺失系和缺體-四體系成功克隆了馴化基因Q,定位中用只在5A染色體Q位點有差異的親本構建了462個F2個體就將該基因成功克隆(Faris等,2003)。但這種方法所開發的分子標記的數量有限,定位精度較差,因此應用有限。
第二種方法是基於比較基因組學的方法。雖然禾本科物種在基因組大小上差異很大,但基因在染色體上的順序保持著廣泛的保守性,即近緣物種間保守的基因共線性關係,這反映了禾本科物種在五千萬年前從同一個祖先種分化而來的事實(Paterson等,2004)。這同時為利用共線性開發分子標記提供了基礎。模式禾本科物種水稻和短柄草擁有小的基因組且和小麥保持著良好的共線性關係,他們的參考基因組序列已經完成,為小麥基因的圖位克隆提供了理想的參照,故被廣泛運用於小麥分子標記的開發當中(Kellogg 2015),如抗條銹病基因Lr67的圖位克隆中利用與水稻和短柄草的保守共線性關係快速的找到了目的基因所在的BAC(Moore等,2015),其他很多基因如Sr33、Sr35等都利用了比較基因組學開發分子標記(Periyannan等,2013;Saintenac等,2013)。當定位區間對應的水稻或短柄草基因組區段得到確定後,即可利用共線性區域內的短柄草或水稻基因開發小麥的探針甚至篩選BAC,這提高了基因圖位克隆的效率,當前的大量數據分析表明65%左右的短柄草或水稻基因和小麥保持著良好的共線性關係(Brutnell等,2015;Kellogg 2015)。但比較基因組學也有一定的局限性,在一些小麥基因組區段發生了重組破壞了和水稻或短柄草的共線性關係,小麥基因組的擴增增加了很多非共線性的基因(Glover等,2015;International Wheat Genome Sequencing Consortium 2014;Wicker等,2010;Wicker等,2011)。如快速進化的抗病基因常分布在重組率較高的基因組區域,此類區域因重組頻繁發生共線性容易遭到了破壞,一些基因組區段甚至找不到對應的水稻或短柄草共線性區間(Leister 2004)。因此禾本科物種間保守的共線性關係為小麥基因的定位提供了參考,但在小麥基因的精細定位、候選基因鑑定中作用有限,特別是在基因組複雜的區段內。
第三種方法是基於晶片技術的方法。基於晶片雜交的分型方法通量高、成本低,在當前挖掘多態性和關聯分析中應用廣泛。當前有小麥9k(Cavanagh等,2013)、90k(Wang等,2014)和820k(Wilkinson等,2012)晶片,標記密度逐漸增高。然而晶片中的標記是依據特定品種開發而來,對於其他品種的分型效率不高,小麥基因的多拷貝特徵使有大量的SNP無信號或並不能分型,同時SNP密度在染色體上分布不均一,大量SNP聚集在少量位點上,另外,其SNP數量固定且定位精度不高,如要對每個個體進行分型則成本較高。因此當前晶片技術雖然可以挖掘大量的變異,但其成本較高且定位精度有限,特別是在小麥重要基因的精細定位和克隆中應用有限。
第四種方法是基於下一代測序技術的方法。下一代測序技術主要以高通量低成本著稱,這些特點大大拓展了可研究的範圍,比如除了得到DNA序列信息之外,我們可以用這些基於測序的方法來研究基因的表達,高效且準確的獲得基因的表達量、稀有轉錄本、選擇性剪切事件、非編碼轉錄本、非編碼區、結構變異和單核苷酸多態性等各種信息,這是基因晶片等技術無法比擬的,實際上基於測序的方法正在取代基因晶片成為研究基因表達最有力的工具(Farnham 2009;Licatalosi等,2010;Wang等,2009)。然而,小麥缺乏參考基因組序列,這大大限制了下一代測序技術在小麥重要基因挖掘和定位中的應用。當前可用的方法是利用GBS(Genotyping By Sequencing)技術,其不依賴於參考基因組序列,利用序列的相似性進行聚類和分型,在小麥中有少量應用報導(Edae等,2015;Mascher等,2013)。但該技術獲得高質量SNP標記有限,且小麥基因的高拷貝特徵容易帶來誤差,其需要對每個個體進行測序成本較高,因而應用有限。
技術實現要素:
本發明的目的是提供一種不依賴於參考基因組序列、低成本、快速、精度高的小麥BSR-Seq基因定位的方法。
本發明的目的是通過以下技術方案實現的:
本發明的小麥BSR-Seq基因定位的方法,包括步驟:
A、混池的構建和測序:
根據重組自交系作圖群體、加倍雙單倍體(DH)群體、回交滲入系群體、F2或F2:3分離群體表型鑑定結果,分別用15-30個以上純合極端高值個體和15-30以上個純合極端低值個體分別組建高值混合池和低值混合池,在表型未表現出差異,或表現出差異後分別取等量葉片組織混合而成高值池和低值池,並提取高值池和低值池的mRNA後進行轉錄組測序,從而得到兩個混池的轉錄組測序數據。
B、高質量變異挖掘:
首先,對轉錄組測序原始數據進行過濾得到高質量數據,過濾標準是去除兩端測序質量值小於20的鹼基,小於25bp的測序讀長將被丟棄,過濾採用自寫Perl程序執行;
其次,用STAR軟體將高質量轉錄組測序序列數據比對到參考序列上並進行過濾,保留只有唯一比對位置且錯配數小於2%的序列比對結果,比對結果使用Samtools軟體挖掘可能的變異位點,再用自寫Perl程序僅保留比對質量大於phred值15、變異質量大於phred值30、只有2種基因型、總深度大於6小於100000、參考序列基因型深度大於3、變異基因型深度大於3、參考序列基因型深度比例大於5%和變異基因型深度比例大於5%的比對結果;
C、與目的基因緊密連鎖的轉錄本的篩選:
混池篩選和目的基因緊密連鎖轉錄本的原理是:和目的基因越近的轉錄本在兩混池間的等位基因頻率差異越大,從而通過計算轉錄本SNP等位基因頻率差異大小可以判斷其與目的基因的遠近;
用自寫Perl腳本從比對結果中得到SNP位點不同基因型在混池中的表達深度,以此計算等位基因頻率;
另外用自寫Perl腳本計算各轉錄本各SNP位點最可能的兩基因型在高值池和低值池的等位基因頻率並計算其差值,同時用Fish精確檢驗計算兩基因型在兩混池中的表達量列聯表差異p-value,排除兩混池間等位基因頻率差值小於0.6和Fish精確檢驗p-value值大於1e-8的SNP位點,然後排除含有兩混池間等位基因頻率差值小於0.6或Fish精確檢驗p-value值大於1e-8的SNP位點的轉錄本,最後剩下的轉錄本我們認為是和目的基因緊密連鎖的轉錄本;
D、分子標記開發和定位:
首先,依據得到的SNP位點設計CAPS或dCAPS標記,並依據與IWGSC資料庫比對的結果找出轉錄本中在A/B/D同源基因間存在差異的特定位置,根據該位置設計EST標記,此外依據轉錄本序列和比對上的IWGSC序列設計SSR標記;
其次,在作圖群體中對分子標記進行多態性檢驗和分型;
最後,依據表型和各標記基因型數據進行遺傳定位。
由上述本發明提供的技術方案可以看出,本發明實施例提供的小麥BSR-Seq基因定位的方法,不依賴於參考基因組序列、低成本、快速、精度高。
附圖說明
圖1為本發明實施例中小麥抗白粉病基因PmTm4混池轉錄組測序高質量變異分布;
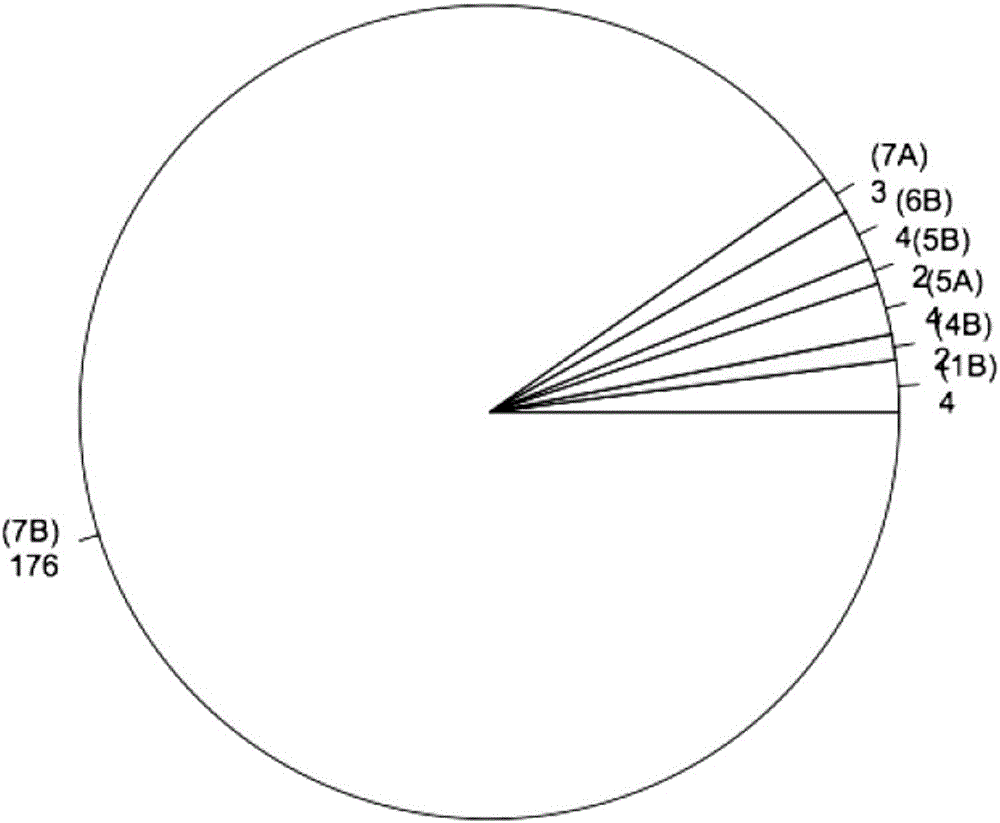
圖2為本發明實施例中小麥抗白粉病基因PmTm4混池轉錄組測序候選SNP組成餅圖;
圖3為本發明實施例中小麥抗白粉病基因PmTm4的遺傳圖譜。
圖3中染色體左邊數字顯示遺傳圖譜上標記的相對位置,染色體右邊表示標記名稱,和PmTm4最近的非共分離標記只存在有一個交換,Xwggc開頭的標記為混池轉錄組測序數據分析而來的分子標記。
具體實施方式
下面將對本發明實施例作進一步地詳細描述。
本發明的小麥BSR-Seq基因定位的方法,其較佳的具體實施方式是:
包括步驟:
A、混池的構建和測序:
根據重組自交系作圖群體、加倍雙單倍體(DH)群體、回交滲入系群體、F2或F2:3分離群體表型鑑定結果,分別用15-30個以上純合極端高值個體和15-30以上個純合極端低值個體分別組建高值混合池和低值混合池,在表型未表現出差異,或表現出差異後分別取等量葉片組織混合而成高值池和低值池,並提取高值池和低值池的mRNA後進行轉錄組測序,從而得到兩個混池的轉錄組測序數據。
B、高質量變異挖掘:
首先,對轉錄組測序原始數據進行過濾得到高質量數據,過濾標準是去除兩端測序質量值小於20的鹼基,小於25bp的測序讀長將被丟棄,過濾採用自寫Perl程序執行;
其次,用STAR軟體將高質量轉錄組測序序列數據比對到參考序列上並進行過濾,保留只有唯一比對位置且錯配數小於2%的序列比對結果,比對結果使用Samtools軟體挖掘可能的變異位點,再用自寫Perl程序僅保留比對質量大於phred值15、變異質量大於phred值30、只有2種基因型、總深度大於6小於100000、參考序列基因型深度大於3、變異基因型深度大於3、參考序列基因型深度比例大於5%和變異基因型深度比例大於5%的比對結果;
C、與目的基因緊密連鎖的轉錄本的篩選:
混池篩選和目的基因緊密連鎖轉錄本的原理是:和目的基因越近的轉錄本在兩混池間的等位基因頻率差異越大,從而通過計算轉錄本SNP等位基因頻率差異大小可以判斷其與目的基因的遠近;
用自寫Perl腳本從比對結果中得到SNP位點不同基因型在混池中的表達深度,以此計算等位基因頻率;
另外用自寫Perl腳本計算各轉錄本各SNP位點最可能的兩基因型在高值池和低值池的等位基因頻率並計算其差值,同時用Fish精確檢驗計算兩基因型在兩混池中的表達量列聯表差異p-value,排除兩混池間等位基因頻率差值小於0.6和Fish精確檢驗p-value值大於1e-8的SNP位點,然後排除含有兩混池間等位基因頻率差值小於0.6或Fish精確檢驗p-value值大於1e-8的SNP位點的轉錄本,最後剩下的轉錄本我們認為是和目的基因緊密連鎖的轉錄本;
D、分子標記開發和定位:
首先,依據得到的SNP位點設計CAPS或dCAPS標記,並依據與IWGSC資料庫比對的結果找出轉錄本中在A/B/D同源基因間存在差異的特定位置,根據該位置設計EST標記,此外依據轉錄本序列和比對上的IWGSC序列設計SSR標記;
其次,在作圖群體中對分子標記進行多態性檢驗和分型;
最後,依據表型和各標記基因型數據進行遺傳定位。
本發明的小麥BSR-Seq基因定位的方法,不依賴於參考基因組序列、低成本、快速、精度高。
本發明將下一代轉錄組測序技術(轉錄組測序,RNA-Seq)和混池技術(Bulked Segregant Analysis,BSA)相結合解決相關問題。首先,利用小麥測序草圖序列(International Wheat Genome Sequencing Consortium 2014)作為參考序列,雖然其基因組覆蓋度約60%但其基因覆蓋度可達到90%,解決了小麥無完整參考轉錄本序列可用問題。其次,採用下一代測序技術高通量挖掘轉錄本上的大量的高質量SNP遺傳變異,再結合混池技術精確計算等位基因頻率來快速的篩選出可能與目的性狀緊密連鎖的轉錄本,並通過Fish精確檢驗控制假陽性。這極大的提升了小麥基因定位的效率和精度並極大的降低了小麥多態性分子標記開發的成本,使小麥基因的精細定位工作時長從數年降低到數月、定位精度從數cM降低到零點幾或0cM以及精細定位成本從數萬降低到數千。本發明對不同小麥性狀相關基因的精細定位和克隆具有重要意義。
具體步驟:
第一,根據重組自交系作圖群體、加倍雙單倍體(DH)群體、回交滲入系群體、F2或F2:3分離群體表型鑑定結果,分別用15-30個以上純合極端高值個體和15-30以上個純合極端低值個體分別組建高值混合池和低值混合池,在表型未表現出差異,或表現出差異後分別取等量葉片組織混合而成高值池和低值池,並提取高值池和低值池的mRNA後進行轉錄組測序,從而得到兩個混池的轉錄組測序數據。
第二,高質量變異挖掘。為了挖掘高質量變異,首先對轉錄組測序原始數據進行過濾得到高質量數據,過濾標準是去除兩端測序質量值小於20的鹼基,小於25bp的測序讀長將被丟棄,過濾採用自寫Perl程序執行;其次,用軟體STAR(Dobin等,2013)將高質量轉錄組測序序列數據比對到參考序列上並進行過濾,保留只有唯一比對位置且錯配數小於2%的序列比對結果。比對結果使用軟體Samtools(Li等,2009)挖掘可能的變異位點,再用自寫Perl程序僅保留比對質量大於phred值15、變異質量大於phred值30、只有2種基因型、總深度大於6小於100000、參考序列基因型深度大於3、變異基因型深度大於3、參考序列基因型深度比例大於5%和變異基因型深度比例大於5%的比對結果。
第三,與目的基因緊密連鎖的轉錄本的篩選。混池篩選和目的基因緊密連鎖轉錄本的原理是,和目的基因越近的轉錄本在兩混池間的等位基因頻率差異越大,從而通過計算轉錄本SNP等位基因頻率差異大小可以判斷其與目的基因的遠近。用自寫Perl腳本從比對結果中得到SNP位點不同基因型在混池中的表達深度,以此計算等位基因頻率。另外用自寫Perl腳本計算各轉錄本各SNP位點最可能的兩基因型在高值池和低值池的等位基因頻率並計算其差值,同時用Fish精確檢驗計算兩基因型在兩混池中的表達量列聯表差異p-value,排除兩混池間等位基因頻率差值小於0.6和Fish精確檢驗p-value值大於1e-8的SNP位點,然後排除含有兩混池間等位基因頻率差值小於0.6或Fish精確檢驗p-value值大於1e-8的SNP位點的轉錄本,最後剩下的轉錄本我們認為是和目的基因緊密連鎖的轉錄本。
第四,分子標記開發和定位。首先依據得到的SNP位點設計CAPS或dCAPS標記,並依據與IWGSC資料庫比對的結果找出轉錄本中在A/B/D同源基因間存在差異的特定位置,根據該位置設計EST標記,此外依據轉錄本序列和比對上的IWGSC序列設計SSR標記。其次在作圖群體中對分子標記進行多態性檢驗和分型。最後依據表型和各標記基因型數據進行遺傳定位。
通過這些步驟,我們成功利用小麥測序草圖作為參考轉錄本序列,並採用嚴格過濾步驟一定程度解決了小麥基因多拷貝帶來的變異挖掘假陽性問題,得到了高質量變異;再利用混池原理,通過等位基因頻率差異來判斷連鎖的轉錄本或變異,並結合Fish精確檢驗有效排除連鎖假陽性的轉錄本。從而得到了有效的、低成本、快速和高定位精度的小麥混池轉錄組測序基因定位技術。
附表:
表1小麥抗白粉病基因PmTm4混池轉錄組測序數據的質量控制結果統計
表2小麥抗白粉病基因PmTm4混池轉錄組測序高質量數據比對結果統計
具體實施例:
實施例一:小麥抗白粉病基因PmTm4的混池轉錄組測序數據分析和精細定位
具體方法為:
(1)混池的構建和測序。為了對小麥抗白粉病基因PmTm4進行精細定,以抗病親本唐麥4號和感病親本農大015為親本組合構建了包含1,504個個體的F2分離群體,並獲得其F2:3家系,表型鑑定後60個純合抗病F2:3家系和60個純合感病F2:3家系在侵染白粉菌後3葉期對每個家系進行取樣,每個家系中的一個個體的第3片葉頂端5釐米(cm)的葉片組織被採集後進行混合,抗病家系混合成抗病混池,感病家系混合成感病混池,並對混池進行RNA提取和轉錄組雙末端測序。抗病混池的數據量為100bp長的73,229,327對Read,感病混池的數據量為100bp長的90,218,629對Read。
(2)高質量變異挖掘。通過質量控制,抗病池和感病池轉錄組測序數據兩條雙末端Read都保留下來的比例超過99%,顯示測序數據質量很高,每個樣本過濾後的數據總量在15Gb左右(表1)。序列比對和過濾後發現,能比對到參考序列上的Read對數大於90%,這說明參考轉錄本序列的完整性較高;比對到唯一位置的可信比對Read對數佔比近70%,比對到多個位置的Read對數佔比在23%左右,因序列差異較大無法比對到參考序列上的Read對數佔比0.39%,這顯示測序數據和參考序列較為相似,且小麥基因組中存在大量高度相似的基因並在過濾中被排除。另外,SNP和Indel發生的概率小於0.15%和0.02%,進一步說明了編碼區測序數據和參考基因組序列的相似性;發現超過2千6百萬個剪切位點,表明小麥基因組基因數可能超過預期,特別是蛋白編碼基因,也說明小麥基因的剪切變體非常豐富(表2)。在抗感池轉錄組數據間找到SNP 256,247個,高質量SNP 106,487個,高質量SNP在各染色體上的數量和各染色體大小成正比(圖1)。
(3)與目的基因緊密連鎖的轉錄本的篩選。計算等位基因頻率差異(AFD)和Fish精確檢驗後,發現關聯的SNP位點主要位於7BL染色體臂上,這和以前的定位結果一致(Hu等,2008)。經過篩選(AFD>0.8,P-value<1e-10),尋找到195個候選SNP,其中176(90)個位於7BL上(圖2),這些SNP集中在對應於短柄草1Mb區域內,這表示篩選效果很好且PmTm4很可能位於該區域內。
(4)分子標記開發和定位。選取15個候選SNP進行分子標記開發,其中11個具有預期的多態性,顯示挖掘出的變異質量較高。對193個F2:3家系的重組個體進行分型,最終PmTm4基因被定為在一個0.51cM的區間內,對應1.9Mb的區間,最近的上下遊非共分離標記都只存在一個交換,極大的改善了該基因的定位精度(圖3)。這些結果表明通過對混池轉錄組數據進行分析尋找到的候選SNP和目的基因緊密連鎖。
以上所述,僅為本發明較佳的具體實施方式,但本發明的保護範圍並不局限於此,任何熟悉本技術領域的技術人員在本發明披露的技術範圍內,可輕易想到的變化或替換,都應涵蓋在本發明的保護範圍之內。因此,本發明的保護範圍應該以權利要求書的保護範圍為準。