氯化氫/乙醚溶液催化製備PCL‑PDMS‑PCL穩定劑的方法與流程
2023-12-03 02:01:31 2
本發明涉及高分子聚合技術領域,具體是氯化氫/乙醚溶液催化製備pcl-pdms-pcl穩定劑的方法。
背景技術:
超臨界二氧化碳(scco2)技術已成為眾多學者研究的熱門課題,由於其具有來源廣泛、有類似氣體的擴散性和液體的密度、無毒、惰性、反應產物易分離純化等優點,使其作為一種綠色溶劑代替了許多有毒有害的有機溶劑而被廣泛的研究和應用,尤其是在分散聚合領域也得以推廣和使用。
超臨界二氧化碳能溶解大多數低分子量的非極性分子和一些極性分子,但大多數工業上應用廣泛的聚合物在較溫和的條件下卻不能被溶解,只有無定型的含氟聚合物和矽氧烷聚合物能完全溶於超臨界二氧化碳,因此,大多數在超臨界二氧化碳中的聚合反應是非均相的,即沉澱聚合。沉澱聚合存在一些缺點,如轉化率低,產物分子量較小以及產物形態不規則等。而分散聚合能在很大程度上克服這些缺點,在穩定劑的作用下,能夠在聚合物與溶劑界面的形成一定的作用力,通過物理吸附或化學接枝產生位阻效應來防止顆粒的凝聚,分散聚合能提高反應效率和收率。分散聚合的效果很大程度取決於分散穩定劑的作用效果。目前製備分散穩定劑多採用金屬催化劑,採用金屬催化劑的製備方法反應時間較長,一般需要24~48h,而且有毒副作用,因此開發綠色環保、成本低廉和錨固效果好的穩定劑是非常重要的。
技術實現要素:
本發明的目的是提供一種用氯化氫乙醚溶液催化製備pcl-pdms-pcl穩定劑的方法。
為實現上述發明目的,本發明的技術方案如下:
氯化氫/乙醚溶液催化製備pcl-pdms-pcl穩定劑的方法,以羥丙基封端的聚二甲基矽氧烷(htpdms)為引發劑,以氯化氫/乙醚溶液(hcl·et2o)為催化劑,引發ε-己內酯(ε-cl)開環聚合,製備三嵌段穩定劑聚己內酯-b-聚二甲基矽氧烷-b-聚己內酯(pcl-pdms-pcl),合成路線如下:
上述方法包括以下步驟:
(1)將htpdms、二氯甲烷、ε-cl加入到燒瓶內,然後加入hcl·et2o溶液,在15~35℃下攪拌3~5小時,並氬氣保護;其中hcl·et2o與htpdms的摩爾比為2~6:1,htpdms與ε-cl單體質量比為1:1~2;
(2)步驟(1)的反應結束後,加入300ml冷甲醇,沉澱析出產物後過濾,再真空乾燥,即得到pcl-pdms-pcl穩定劑,反應路線如下:
優選地,由於htpdms鏈的兩端分別含有一個起到引發作用的羥基,根據引發機理選擇hcl·et2o與htpdms的摩爾比為2:1。
優選地,步驟(1)中反應溫度為25℃,攪拌4h。
合成機理:氯化氫/乙醚溶液催化劑的陽離子與ε-己內酯生成氧鎓正離子,己內酯的羰基被活化,然後,聚二甲基矽氧烷兩端的羥基進攻活性羰基,促使己內酯開環聚合,醯氧鍵斷裂,反應原理路線見圖1。
與現有技術相比,本發明的優點在於:本發明採用氯化氫/乙醚溶液為催化劑,反應條件比較溫和,避免了使用金屬催化劑的毒副作用,製備環境更加綠色,反應時間明顯縮短,製備的三嵌段穩定劑pcl-pdms-pcl其分子量可達6384g/mol,產率最高可達到86%以上,該穩定劑可用於超臨界流體中生物醫藥材料聚合的分散劑,可用於超臨界二氧化碳中生物材料聚乳酸、聚己內酯及其共聚物等脂肪族聚酯的合成及其它功能材料的合成。
附圖說明
圖1本發明合成原理圖;
圖2pcl-pdms-pcl紅外光譜圖;
圖3pcl-pdms-pcl核磁共振氫譜譜圖。
具體實施方式
下面結合具體實施例對本發明做進一步說明。
實施例1
將稱量好的htpdms5g(0.0015mol),乾燥除水的二氯甲烷30ml,ε-cl5g(0.044mol)加入到50ml三口圓底燒瓶內,然後加入4.2mol/l的hcl·et2o溶液0.71ml(0.003mol),25℃下攪拌4小時,並氬氣保護;反應結束後,加入300ml冷甲醇中,沉澱析出產物後過濾,經真空乾燥得到產品pcl-pdms-pcl8.6g,分子量為6384g/mol,產率為86%。
實施例2
將稱量好的htpdms5g(0.0015mol),乾燥除水的二氯甲烷30ml,ε-cl5g(0.044mol)加入到50ml三口圓底燒瓶內,然後加入4.2mol/l的hcl·et2o溶液1.42ml(0.006mol),25℃下攪拌4小時,並氬氣保護;反應結束後,加入300ml冷甲醇中,沉澱析出產物後過濾,經真空乾燥得到產品pcl-pdms-pcl8.5g,分子量為6016g/mol,產率為85%。
實施例3
將稱量好的htpdms5g(0.0015mol),乾燥除水的二氯甲烷30ml,ε-cl5g(0.044mol)加入到50ml三口圓底燒瓶內,然後加入4.2mol/l的hcl·et2o溶液0.71ml(0.003mol),35℃下攪拌4小時,並氬氣保護;反應結束後,加入300ml冷甲醇中,沉澱析出產物後過濾,經真空乾燥得到產品pcl-pdms-pcl7.8g,分子量為6023g/mol,產率為78%。
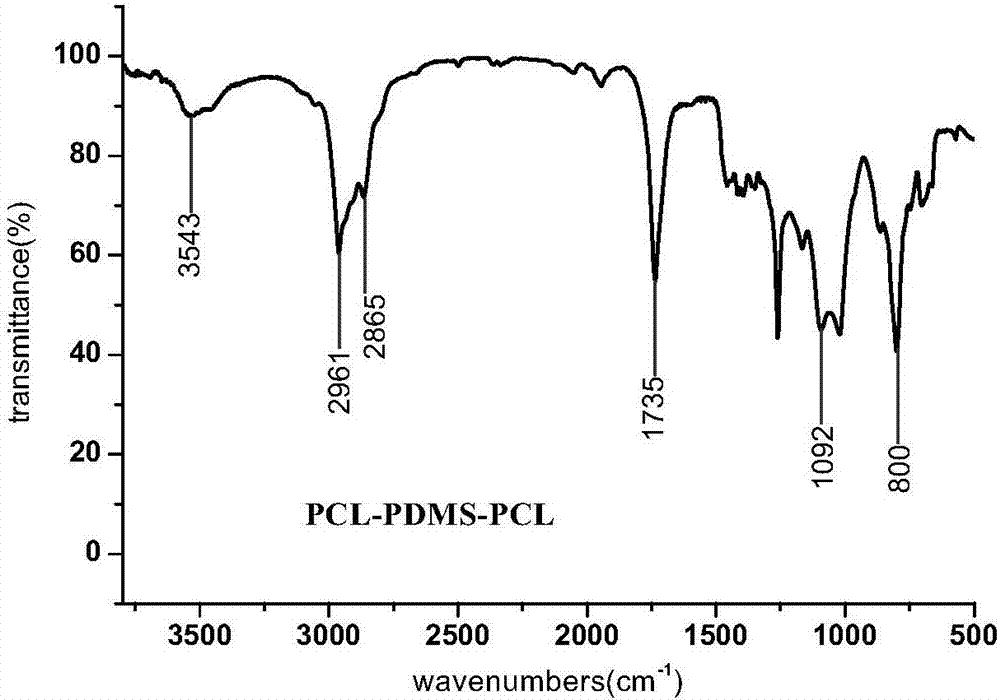
圖2為實施例1中得到的三嵌段穩定劑pcl-pdms-pcl的紅外光譜圖。在3543cm-1處為產物端羥基(-oh)的伸縮振動峰,在2961cm-1和2865cm-1處為pcl-pdms-pcl中pcl的亞甲基的c-h伸縮振動峰,在1735cm-1處為pcl中羰基(c=o)的伸縮振動峰,在800cm-1處為pdms中si-c鍵的伸縮振動峰,在1092cm-1處為pdms主鏈中si-o鍵的伸縮振動峰。上述結果表明,譜圖中的吸收峰符合pcl-pdms-pcl的特徵吸收峰。
圖3為實施例1中得到的三嵌段穩定劑pcl-pdms-pcl的1h-nmr譜圖。1hnmr(cdcl3,ppm):4.04–4.07[t,cooch2ch2,ha+e];2.28–2.32[t,ch2ch2coo,hd];1.60–1.68[m,cooch2ch2ch2andch2ch2ch2coo,hb+f];1.35–1.41[m,ooch2ch2ch2,hc];0.48–0.55[t,ch2ch2ch2si,hg];0.03–0.09[m,si(ch3)2,hh]。其中,0.05ppm和0.5ppm處的峰為聚二甲基矽氧烷的特徵峰,1.7ppm和2.3ppm為己內酯重複單元的特徵信號,證明了聚合物為三嵌段穩定劑pcl-pdms-pcl。
由紅外光譜圖2和核磁共振圖3分析表明,經氯化氫/乙醚溶液催化合成的三嵌段穩定劑pcl-pdms-pcl的結構與設計的目標分子結構一致。
用氯化氫/乙醚溶液作為催化劑,在製備pcl-pdms-pcl過程中只起到活化劑的作用,不屬於引發劑;該方法反應時間明顯縮短,比辛酸亞錫催化反應時間快20h;製備環境更加綠色,是一種具有發展潛力的穩定劑的合成方法,符合綠色化學發展的方向,有著廣闊的應用前景。