橙花醇脂肪酸酯類衍生物及其應用和製備方法與流程
2023-05-05 06:44:26 2
本發明涉及一種衍生物及其衍生物的應用和製備方法,尤其是一種橙花醇脂肪酸酯類衍生物及其應用和製備方法。
背景技術:
經皮給藥系統(transdermaldrugdeliverysystems,tdds)是藥物經過皮膚吸收,進入體循環而產生療效的一類新製劑。tdds一經出現,就以其持久、恆定和可控的血藥濃度,避免肝首過效應,給藥方便,患者依從性高等優點倍受醫藥界的關注。皮膚是防止外界物質進入體內和體內水分散失的天然屏障。皮膚由表皮、真皮和皮下組織構成,其中表皮又可分為角質層和活性表皮,角質層是藥物經皮吸收的主要屏障。大部分藥物經皮給藥後,透過速率遠遠達不到治療要求,因此,為了使更多的藥物開發成tdds,尋找增加藥物透過量的方法是tdds研究的當務之急。應用促透劑的化學促透法是最古老的方法,但它卻是最簡單、安全、廉價和實用的方法。萜類促透劑大多來源於天然產物(揮髮油),具有促透活性強,毒性低,刺激性小,對親水性和親脂性藥物均有促透作用等優點。但包括萜品醇在內的萜類化合物的揮發性,會給經皮吸收製劑的生產及貯存帶來障礙。由於它易揮發而會造成在製劑中的含量不穩定,而且導致批次間的重現性也較差。常用的萜類促透劑如l-薄荷醇、α-萜品醇、橙花醇等,l-薄荷醇申請者已對其進行了結構改造,製備了17種衍生物,並從中篩選出了較l-薄荷醇低毒、高效、不揮發的促透劑(ligangzhao,liangfang,yongnanxu,etal.transdermaldeliveryofpenetrantswithdifferinglipophilicitiesusingo-acylmentholderivativesaspenetrationenhancers.eurjpharmbiopharm,2008,69:199–213;ligangzhao,liangfang,yongnanxu,etal.effectofo-acylmentholontransdermaldeliveryofdrugswithdifferentlipophilicity.intjpharm,2008,352:92–103),並申請了專利(專利證書號:zl200710158246.1)。
橙花醇是一種對人體有益的揮髮油類物質,同時它還能促鹽酸阿夫唑嗪(dprasanthi,pklakshmi.terpenes:effectoflipophilicityinenhancingtransdermaldeliveryofalfuzosinhydrochloride.jadvpharmtechnolres,2012,3:216-23)、依美斯汀(sharada,ynakada,fyamashita,etal.biopharmaceuticalconsiderationsonantihistamineeffectsoftopicallyadministeredemedastine.jpharmsci,2005,94:17-24.)等多種藥物的經皮吸收,也是一種較為常見的天然促透劑,所以本發明人對其衍生物也進行了系統研究。
技術實現要素:
本發明的目的在於提供一種具有促透活性的橙花醇脂肪酸酯類衍生物及在經皮給藥製劑中的應用。
本發明採用如下技術方案:
一種橙花醇脂肪酸酯類衍生物,該衍生物的結構通式如下:
該衍生物的優選方案如下:
橙花醇為天然或合成α-橙花醇(結構式a)、β-橙花醇(結構式b);脂肪酸包括飽和直鏈脂肪酸及不飽和直鏈脂肪酸。
飽和脂肪酸為c2~c18的直鏈脂肪酸或r=c1~c17,不飽和脂肪酸的r為:(ch2)7ch=ch(ch2)7ch3。
飽和脂肪酸包括丙酸、丁酸、己酸、庚酸、辛酸、癸酸、十二酸、十四酸、十六酸和十八酸;不飽和脂肪酸為油酸。
一種橙花醇脂肪酸酯類衍生物的應用,作為經皮吸收促透劑應用,製備經皮給藥製劑,提高藥物的經皮吸收,增加藥物的累積透過量。
橙花醇脂肪酸酯類衍生物的應用的優選方案是:
經皮給藥製劑分別是貼劑、巴布劑、乳膠劑、凝膠劑、乳膏劑、軟膏劑、搽劑或噴霧劑類外用製劑。
製劑的製備方法如下:將藥物成分、壓敏膠和橙花醇脂肪酸酯衍生物充分混合後以轉移性塗布製成於防粘層上,經過30~100℃乾燥,然後用包括pvc或無紡布的裱褙材料複合,即得到經皮給藥製劑。
藥物成分選自雙氯芬酸鉀、吲哚美辛、酮洛芬、奧昔布寧、氟比洛芬、比索洛爾、利多卡因、司來吉蘭或硝酸異山梨酯中的一種。
壓敏膠是選自矽酮壓敏膠、丙烯酸酯壓敏膠或聚異丁烯壓敏膠中的一種。
橙花醇脂肪酸酯衍生物的有效用量為0.5%-20%(w/w)。
橙花醇脂肪酸酯衍生物的有效用量為3%-10%(w/w)。
將壓敏膠中加入雙氯芬酸鉀後,攪拌直至藥物完全溶於壓敏膠溶液;之後,加入橙花醇脂肪酸酯衍生物及甘油、乙酸乙脂和羥丙基纖維素,繼續攪拌,直至形成澄清黏稠狀溶液;隨後,採用實驗用塗布機轉移塗布於防粘層上,經30℃~100℃乾燥後,複合背襯無紡布,即得雙氯芬酸鉀貼劑。
一種橙花醇脂肪酸酯類衍生物的製備方法,脂肪酸通過與氯化亞碸反應後得到脂肪醯氯,脂肪醯氯與橙花醇進行反應。
橙花醇脂肪酸酯類衍生物的製備方法的優選方案是:
製備過程中所用溶劑為除去水的低毒有機溶劑。
橙花醇為天然或合成α-橙花醇(結構式a)、β-橙花醇(結構式b);脂肪酸包括飽和直鏈脂肪酸及不飽和直鏈脂肪酸。
收集不同化學結構的脂肪酸,按照如下所示的合成路線,合成橙花醇酯類衍生物;化合物1-11,均採如下用路線合成:
本發明所合成的上述橙花醇脂肪酸類衍生物可作為經皮吸收促透劑應用,製備經皮給藥製劑(貼劑、巴布劑、乳膠劑、乳膏劑、凝膠劑、軟膏劑、搽劑、噴霧劑等),也可利用衍生物的低揮發性作為橙花醇替代品而用於含有橙花醇的外用製劑中。
本發明所述的化合物可應用於經皮給藥製劑中,增強藥物的透過能力,也可作為香料,掩蓋製劑的不良氣味,有廣泛的潛在應用前景。製備方法適合於工業化生產,安全,易於保存。
附圖說明
圖1為:應用於貼劑中橙花醇脂肪酸酯促進雙氯芬酸鉀的透皮吸收累積透過量圖。
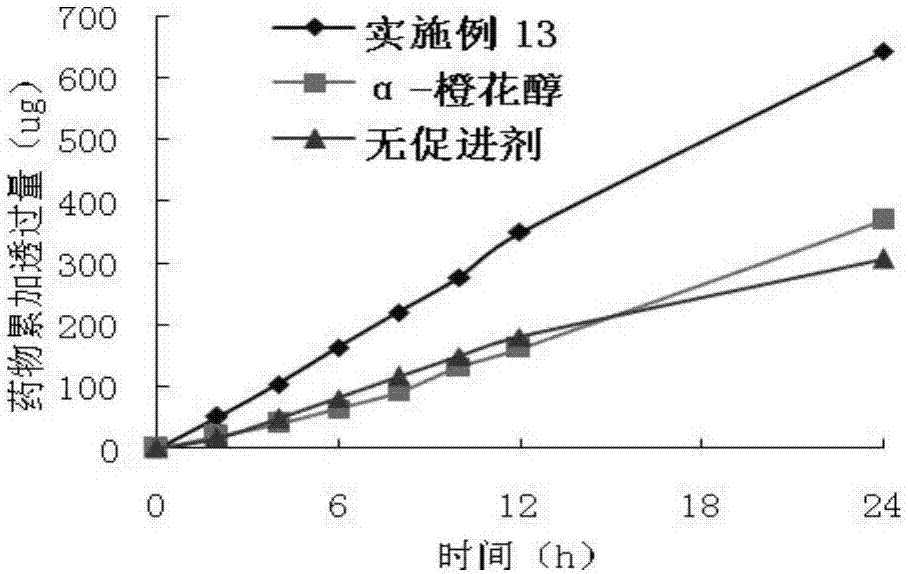
圖2為:應用於貼劑中橙花醇脂肪酸酯促進吲哚美辛的透皮吸收累積透過量圖。
圖3為:應用於貼劑中橙花醇脂肪酸酯促進酮洛芬的透皮吸收累積透過量圖。
圖4為:應用於貼劑中橙花醇脂肪酸酯促進奧昔布寧的透皮吸收累積透過量圖。
圖5為:應用於貼劑中橙花醇脂肪酸酯促進吲氟比洛芬的透皮吸收累積透過量圖。
圖6為:應用於貼劑中橙花醇脂肪酸酯促進比索洛爾的透皮吸收累積透過量圖。
圖7為:應用於貼劑中橙花醇脂肪酸酯促進利多卡因的透皮吸收累積透過量圖。
圖8為:應用於貼劑中橙花醇脂肪酸酯促進司來吉蘭的透皮吸收累積透過量圖。
圖9為:應用於貼劑中橙花醇脂肪酸酯促進硝酸異山梨酯的透皮吸收累積透過量圖。
具體實施方式
下面結合附圖及實施例對本發明做進一步的詳細描述:
實施例1:
取丙酸14.8g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於60℃反應3小時,加入含有15.4g(0.1mol)的α-橙花醇四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體,產率為:94%。產物的1hnmr和ms數據如下:esi-msm/z:211.3[m+1]+;1hnmr(cdcl3),:0.90(3h,s),1.01(3h,s),0.99(3h,s),1.25(3h,s),1.30~1.35(2h,m),1.41(1h,m),1.58~1.60(2h,m),1.88(1h,m),2.03(2h,m),5.20(2h,d,j=7.2hz),6.58(1h,d,j=11.2hz)。
實施例2:
取丁酸17.6g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於60℃反應3小時,加入含有15.4g(0.1mol)α-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體20.91g,率為:92.4%。產物的1hnmr和ms數據如下:esi-msm/z:225.3[m+1]+;1hnmr(cdcl3):0.90(3h,s),1.01(3h,s),0.99(3h,s),1.25(3h,s),1.30~1.35(4h,m),1.41(1h,m),1.58~1.60(2h,m),1.88(1h,m),2.03(2h,m),5.20(2h,d,j=7.2hz),6.58(1h,d,j=11.2hz)。
實施例3:
取己酸23.2g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於60℃反應3小時,加入含有15.4g(0.1mol)α-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體23.94g,產率為:93.4%。產物的1hnmr和ms數據如下:esi-msm/z:253.4[m+1]+;1hnmr(cdcl3):0.90(3h,s),1.00(3h,s),1.01(3h,s),1.24(3h,s),1.30~1.35(8h,m),1.40(1h,m),1.58~1.60(2h,m),1.89(1h,m),2.04(2h,m),5.20(2h,d,j=7.2hz),6.58(1h,d,j=11.2hz)。
實施例4:
取庚酸26g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於60℃反應3小時,加入含有15.4g(0.1mol)α-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體24.41g,產率為:91%。產物的1hnmr和ms數據如下:esi-msm/z:267.4[m+1]+;1hnmr(cdcl3):0.91(3h,s),0.99(3h,s),1.01(3h,s),1.23(3h,s),1.30~1.36(10h,m),1.40(1h,m),1.58~1.62(2h,m),1.89(1h,m),2.03(2h,m),5.20(2h,d,j=7.2hz),6.58(1h,d,j=11.2hz)。
實施例5:
取辛酸28.8g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於60℃反應3小時,加入含有15.6g(0.1mol)α-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體259.4g,產率為:90%。產物的1hnmr和ms數據如下:esi-msm/z:281.4[m+1]+;1hnmr(cdcl3):0.91(3h,s),0.99(3h,s),1.01(3h,s),1.23(3h,s),1.30~1.36(10h,m),1.40(1h,m),1.58~1.62(2h,m),1.89(1h,m),2.03(2h,m),5.20(2h,d,j=7.2hz),6.58(1h,d,j=11.2hz)。
實施例6:
取癸酸34.4g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於60℃反應3小時,加入含有15.4g(0.1mol)β-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體30.83g,產率為:93.5%。產物的1hnmr和ms數據如下:esi-msm/z:309.5[m+1]+;1hnmr(cdcl3):1.21(3h,s),1.30(3h,s),1.31(3h,s),1.75~1.87(19h,m),1.40(1h,m),1.58~1.61(2h,m),1.89(1h,m),2.04(2h,m),6.78(1h,d,j=11.2hz)。
實施例7:
取十二酸40g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於70℃反應3小時,加入含有15.4g(0.1mol)β-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體30.62g,產率為:90.5%。產物的1hnmr和ms數據如下:esi-msm/z:337.5[m+1]+;1hnmr(cdcl3):1.21(3h,s),1.30(3h,s),1.31(3h,s),1.75~1.87(23h,m),1.41(1h,m),1.58~1.61(2h,m),1.89(1h,m),2.04(2h,m),6.78(1h,d,j=11.2hz)。
實施例8:
取十四酸45.6g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於70℃反應3小時,加入含有15.4g(0.1mol)β-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得無色液體32.6g,產率為:89%。產物的1hnmr和ms數據如下:esi-msm/z:365.5[m+1]+,1hnmr(cdcl3):1.22(3h,s),1.30(3h,s),1.31(3h,s),1.75~1.87(25h,m),1.42(1h,m),1.58~1.61(2h,m),1.89(1h,m),2.04(2h,m),6.78(1h,d,j=11.2hz)。
實施例9:
取十六酸51.2g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於70℃反應3小時,加入含有15.4g(0.1mol)β-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得白色固體34.3g,產率為:87%。產物的1hnmr和ms數據如下:esi-msm/z:393.6[m+1]+,1hnmr(cdcl3):1.22(3h,s),1.31(3h,s),1.32(3h,s),1.75~1.87(29h,m),1.41(1h,m),1.58~1.61(2h,m),1.89(1h,m),2.04(2h,m),6.78(1h,d,j=11.2hz)。
實施例10:
取十八酸56.8g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於70℃反應3小時,加入含有15.4g(0.1mol)β-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得白色固體35.9g,產率為:85%。產物的1hnmr和ms數據如下:esi-msm/z:421.6[m+1]+;1hnmr(cdcl3):1.22(3h,s),1.31(3h,s),1.32(3h,s),1.75~1.87(33h,m),1.41(1h,m),1.58~1.61(2h,m),1.89(1h,m),2.04(2h,m),6.78(1h,d,j=11.2hz)。
實施例11:
取油酸56.4g(0.2mol),加入氯化亞碸17.85g(0.15mol),混勻後於70℃反應3小時,加入含有15.4g(0.1mol)β-橙花醇的四氫呋喃溶液20ml,繼續反應3小時,反應液用naoh溶液調至ph為6~7,分出有機層,水層用乙酸乙酯提取,合併有機層,用飽和食鹽水洗,無水na2so4乾燥過夜,蒸去溶劑,柱層析(矽膠為200~300目),乙石油醚和乙酸乙酯(15:1)為洗脫劑,蒸除洗脫劑,得微黃色液體36.14g,產率為:84%。產物的1hnmr和ms數據如下:esi-msm/z:419.6[m+1]+;1hnmr(cdcl3):1.22(3h,s),1.31(3h,s),1.32(3h,s),1.75~1.87(33h,m),1.41(1h,m),1.58~1.61(2h,m),1.89(1h,m),2.04(2h,m),6.78(1h,d,j=11.2hz)。
實施例12:在貼劑中橙花醇衍生物促進雙氯芬酸鉀的透皮吸收
皮膚的製備:取體重180g~220g的雄性大鼠,腹腔注射1ml25%的烏拉坦溶液麻醉,剪去腹部毛,處死,取下脫毛皮膚,剝離皮下粘連組織,選完整皮膚用生理鹽水衝洗3次,-20℃冰箱短期保存,備用。
貼劑的製備:在30ml樣品瓶中,裝入丙烯酸酯壓敏膠。在添加雙氯芬酸鉀後,在300rpm下攪拌直至藥物成分完全溶於壓敏膠溶液。之後加入吸收促透劑及其他成分(如下表所示),繼續攪拌,直至形成均一溶液。隨後,採用實驗用塗布機轉移塗布於防粘層上,經30℃~100℃乾燥後,複合背襯無紡布,衝切成7×7cm2大小,即得雙氯芬酸鉀貼劑。
表1
方法:經皮吸收試驗採用水平式雙室擴散池,將製備好的雙氯芬酸鉀貼劑貼敷於處理好的皮膚角質層一側,以ph7.4的磷酸鹽緩衝溶液為接收液,用hplc測定接收池中藥物量,計算累積透過量和透過流通量。其中雙氯芬酸鉀貼劑中促透劑的濃度為(0或5%橙花醇或與5%橙花醇等摩爾濃度)。
實施例13:
除用吲哚美辛代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表2
實施例14:
除用酮洛芬代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表3
實施例15:
除用奧昔布寧代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表4
實施例16:
除用氟比洛芬代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表5
實施例17:
除用比索洛爾代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表6
實施例18:
除用利多卡因代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表7
實施例19:
除用司來吉蘭代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表8
實施例20:
除用硝酸異山梨酯代替雙氯芬酸鉀外,用與實施例12同樣的方法。
表9