一種基於光熱轉換納米材料溫度變化的檢測溶液濃度的方法與流程
2023-05-13 07:35:41 2
本發明屬於分析檢測技術領域,具體涉及一種基於光熱轉換納米材料溫度變化的檢測溶液濃度的方法。
背景技術:
分析檢測技術是用指定的方法檢驗測試氣體、液體或固體指定的技術性能指標。適用於多種行業範疇的質量評定,如環境、食品安全、水質等。目前常見的分析方法有光學檢測、電化學檢測等。但是,目前常見的分析方法存在對儀器要求高,儀器不易攜帶,難以實現可移動式檢測等。
技術實現要素:
本發明的目的在於提供一種基於光熱轉換納米材料溫度變化的檢測溶液濃度的方法,該方法利用光熱轉換納米材料的熱信號的變化實現對待測成分的快速、靈敏、便攜的定量檢測。
本發明所提供的檢測溶液濃度的方法,包括如下步驟:
1)合成含有光熱轉換材料的納米材料,得到光熱轉換納米材料;
2)分別用一系列標準濃度的待測溶液與所述光熱轉換納米材料進行反應,得到一系列混合液,並分別在近紅外光照射下,檢測其在某一時間點處溫度變化,做溫度和濃度的標準曲線;
3)將未知濃度的待測溶液與所述光熱轉換納米材料進行反應,得到混合液,在近紅外光照射下,檢測其在某一時間點處溫度變化,與所述標準曲線對比,即可得到所述待測溶液的濃度。
上述檢測溶液濃度的方法中,步驟1)中,所述光熱轉換材料選自貴金屬,共軛聚合物,有機染料,半導體化合物和碳材料中的至少一種,但不局限於此,其它光熱轉換材料亦適用。
其中,所述貴金屬具體選自金、銀、鉑和鈀等中的至少一種。
所述共軛聚合物具體選自聚吡咯、聚苯胺和聚多巴胺等中的至少一種。
所述有機染料具體選自吲哚菁綠、普魯士藍和滕氏藍等中的至少一種。
所述半導體化合物具體選自CuxS和/或CdSe等,x具體為1-2。
所述碳材料具體選自碳納米管、石墨烯和碳量子點等中的至少一種。
所述光熱轉換納米材料可為僅由光熱轉換材料構成的納米材料或其他納米材料與 光熱轉換材料複合而形成的納米複合材料;
所述光熱轉換納米材料中的光熱轉換材料的質量分數m為0<m≤100%。
所述光熱轉換納米材料可為納米顆粒和/或納米片,其中,所述納米顆粒的直徑為5nm-999nm,所述納米片的邊長為15nm-999nm,厚度為1-15nm。
所述光熱轉換納米材料可通過常規方法製備得到,如:水熱法,溶劑熱法,反相微乳法,熱力學還原法等。
所述光熱轉換納米材料具體可為吲哚菁綠修飾的NaLuF4:Yb,Er納米顆粒、銀包覆的NaGdF4:Yb,Er納米顆粒和三角形銀納米片中的任一種。
其中,所述吲哚菁綠修飾的NaLuF4:Yb,Er納米顆粒中吲哚菁綠的質量分數為0.1-2%。
所述銀包覆的NaGdF4:Yb,Er納米顆粒中銀的包覆厚度為1-10nm,具體可為3nm。
所述三角形銀納米片的邊長為10-500nm,具體可為10-50nm(如:30nm)。厚度為1-10nm,具體可為3nm。
上述檢測溶液濃度的方法中,步驟2)中,所述一系列標準濃度的待測溶液的濃度範圍為250pM-16nM,具體可為250pM、500pM、1nM、2nM、4nM,8nM、16nM。
所述光熱轉換納米材料是以光熱轉換納米材料水溶液的形式參加反應的,所述光熱轉換納米材料水溶液的質量濃度為100μg/mL-10mg/mL,具體可為200μg/mL;
所述光熱轉換納米材料水溶液分別與所述一系列標準濃度的待測溶液的體積比為(0.125-1):1,具體可為0.25:1。
上述檢測溶液濃度的方法中,步驟2)和步驟3)中,所述待測溶液中的待測成分為可與所述光熱轉換納米材料發生絡合和/或氧化還原反應,並使所述光熱轉換納米材料在近紅外區吸收峰減弱的物質。
所述待測溶液具體可為過氧化氫(H2O2)水溶液和抗壞血酸(AA)水溶液中的任一種。
上述檢測溶液濃度的方法中,步驟2)和步驟3)中,所述反應的反應溫度為10-40℃,反應時間為0.5-10min,具體可為5min。
所述某一時間點為從近紅外光照射開始後的時間,所述照射時間為5s-15min,具體可為2min。
上述檢測溶液濃度的方法中,步驟2)和步驟3)中,所述近紅外光的範圍為750-1064nm。
所述近紅外光可優選為如下具體的形式:
當所述光熱轉換納米材料為吲哚菁綠修飾的NaLuF4:Yb,Er水溶液時,所述近紅外 光的波長優選為785nm。
當所述光熱轉換納米材料為銀包覆的NaGdF4:Yb,Er水溶液時,所述近紅外光的波長優選為808nm。
當所述光熱轉換納米材料為三角形銀納米片水溶液時,所述近紅外光的波長優選為980nm。
所述檢測的檢測溫度為1-80℃。
所述溫度變化是通過熱成像分析儀進行的檢測,型號為FLIR-E40。
本發明中步驟2)和步驟3)中的所述反應的反應時間、照射時間、檢測溫度、所述光熱轉換納米材料的加入量和加入濃度應保持一致,保證相同的測試環境,此外,所述待測溶液並不需要完全與所述光熱轉換納米材料反應,而是利用所述光熱轉換納米材料表面的光熱轉換材料與所述待測溶液中的待測成分發生反應,導致光熱轉換材料的減少,引起了溫度的變化,故加入的所述光熱轉換納米材料的量應一致。
本發明運用納米材料的熱信號的變化實現對待測成分的快速、靈敏、便攜的定量檢測。具體通過測得的一系列已知濃度的待測成分的溶液的溫度的值與濃度的線性圖譜(回歸係數R2≥0.99),得到標準線性圖譜。再測未知濃度的待測成分的溫度,與標準線性圖譜對比即可得知。
與現有技術相比,本發明具有如下有益效果:
1)本發明的方法能快速、靈敏對待測液體中的待測組分進行定量分析,提供了一種新的分析測試方法;
2)本發明方法中使用的材料簡單,所需儀器的價格低,攜帶方便,能夠實現低成本的快速、靈敏、便攜的定量分析。
3)本發明分析檢測方法可用於環境、食品、藥品和活體樣品等樣品的檢測。
附圖說明
圖1為實施例1中的吲哚菁綠修飾的NaLuF4:Yb,Er納米顆粒的透射電子顯微鏡照片。
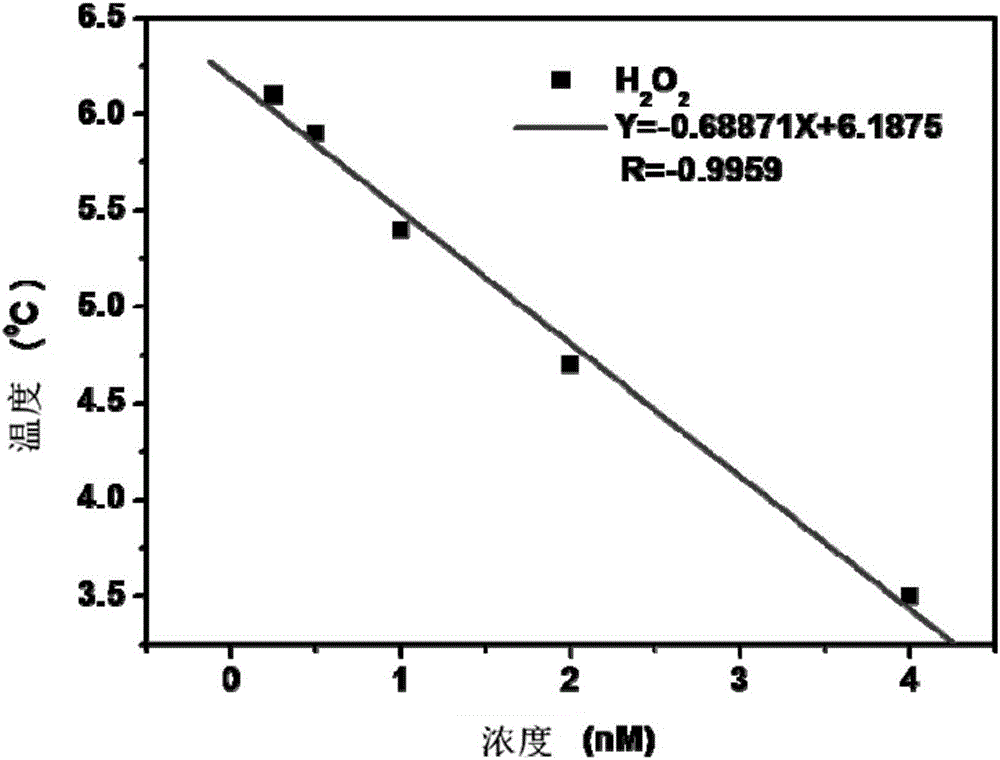
圖2為實施例1中的吲哚菁綠修飾的NaLuF4:Yb,Er納米顆粒檢測過氧化氫(H2O2)的熱標準線性圖譜。
圖3為實施例2中的銀包覆的NaGdF4:Yb,Er納米顆粒的透射電子顯微鏡照片。
圖4為實施例2中的銀包覆的NaGdF4:Yb,Er納米顆粒檢測過氧化氫(H2O2)的熱標準線性圖譜。
圖5為實施例3中的三角形銀納米片的透射電子顯微鏡照片。
圖6為實施例4中的三角形銀納米片檢測抗壞血酸(AA)熱標準線性圖譜。
具體實施方式
下面通過具體實施例對本發明的方法進行說明,但本發明並不局限於此。
下述實施例中所述實驗方法,如無特殊說明,均為常規方法;所述試劑和材料,如無特殊說明,均可從商業途徑獲得。
下述實施例中所用的納米材料NaLuF4:Yb,Er是按照如下方法製備得到:
1)首先,將0.80mmol LuCl3、0.18mmol YbCl3和0.02mmol ErCl3加入到100mL的三口瓶中,再加入6mL油酸和15mL十八烯;然後在氮氣的保護下,將混合溶液加熱到120℃使稀土氯化物完全溶解,形成透明的澄清溶液後,停止加熱,冷卻至室溫;
2)之後,向澄清溶液中加入6mL NaOH(2.5mmol)和NH4F(4mmol)的甲醇溶液,氮氣保護下加熱至80℃除甲醇,約30min後,升溫至120℃抽真空除水除氧;最後在氮氣氛圍下反應1h。反應結束後,自然冷卻至室溫;然後加入適量的環己烷和乙醇,離心分離,去掉上清液;向固體中加入適量環己烷後超聲分散,再加入適量乙醇後,再離心分離;重複以上步驟,繼續用環己烷和乙醇洗滌幾次後,即可得到納米材料NaLuF4:Yb,Er(納米顆粒,直徑為20-28nm)。
下述實施例中所用的納米材料NaGdF4:Yb,Er是按照如下方法製備得到:
1)首先,將0.80mmol GdCl3、0.18mmol YbCl3和0.02mmol ErCl3加入到100mL的三口瓶中,再加入6mL油酸和15mL十八烯;然後在氮氣的保護下,將混合溶液加熱到120℃使稀土氯化物完全溶解,形成透明的澄清溶液後,停止加熱,冷卻至室溫;
2)之後,向澄清溶液中加入6mL NaOH(2.5mmol)和NH4F(4mmol)的甲醇溶液,氮氣保護下加熱至80℃除甲醇,約30min後,升溫至120℃抽真空除水除氧;最後在氮氣氛圍下反應1h。反應結束後,自然冷卻至室溫,然後加入適量的環己烷和乙醇,離心分離,去掉上清液;向固體中加入適量環己烷後超聲分散,再加入適量乙醇後,再離心分離;重複以上步驟,繼續用環己烷和乙醇洗滌幾次後,即可得到納米材料NaGdF4:Yb,Er(納米棒,長度為30nm,直徑為7-9nm)。
下述實施例中所用的吲哚菁綠修飾的NaLuF4:Yb,Er是按照如下方法製備得到:
將質量分數為1mg mL-1的納米材料NaLuF4:Yb,Er的水溶液與NOBF4以質量比1:1混合超聲處理,處理的溫度為20℃,時間為5min,洗去表面的油溶性配體,然後分別用CH2Cl2和無水乙醇洗滌兩遍,再分散在5mL去離子水中。加入1mL 1mM的吲哚菁綠水溶液攪拌反應30min,離心分離,用去離子水洗滌三次,得到吲哚菁綠修飾的 NaLuF4:Yb,Er,其透射電子顯微鏡照片如圖1所示,從圖1可得知:納米材料的尺寸均勻,分散性好。其中,所述吲哚菁綠修飾的NaLuF4:Yb,Er中吲哚菁綠的質量分數為1%。
下述實施例中所用的銀包覆的NaGdF4:Yb,Er是按照下述方法製備得到:
將質量分數為1mg mL-1的納米材料NaGdF4:Yb,Er的水溶液與NOBF4以質量比1:1混合超聲處理,處理的溫度為20℃,時間為5min,洗去表面的油溶性配體,然後分別用CH2Cl2和無水乙醇洗滌兩遍,然後分散在含有質量分數為20%檸檬酸鈉的去離子水中,在30℃下攪拌處理1h。經過檸檬酸處理後,加入5mL 0.5mM的AgNO3水溶液,繼續攪拌5min,然後加入1.5mL 30mM的檸檬酸鈉和10mM的硼氫化鈉溶液,離心分離,用去離子水洗滌三次,得到銀包覆的NaGdF4:Yb,Er,其透射電子顯微鏡照片如圖3所示,從圖3可得知:納米材料的尺寸均勻,分散性好。其中,所得銀包覆的NaGdF4:Yb,Er中銀包覆的厚度為3nm。
下述實施例中所使用的三角形納米銀片是按照下述方法製備得到:
將25mL 0.1mM的AgNO3水溶液、1.5mL 30mM的檸檬酸鈉水溶液、1.5mL 0.7mM的聚乙烯基吡咯烷酮水溶液和60μL 30%的過氧化氫水溶液混合,避光攪拌15min。隨後,滴加250μL 100mM的硼氫化鈉水溶液,繼續避光攪拌反應30min,離心分離,用去離子水洗滌三次,得到三角形納米銀片,其透射電子顯微鏡照片如圖5所示,從圖5可得知:納米材料的尺寸均勻,分散性好,其中,所述三角形納米銀片的邊長為30nm,厚度為3nm。
實施例1、檢測過氧化氫(H2O2)水溶液的濃度:
1)標準曲線的繪製:分別將200μL的250pM、500pM、1nM、2nM、4nM的過氧化氫(H2O2)水溶液與800μL 200μg/mL吲哚菁綠修飾的NaLuF4:Yb,Er水溶液均勻混合,室溫(25℃)靜置反應10min後,反應已完全,在785nm近紅外光照射下測定混合液照射2min後的溫度變化,處理數據得到溫度的線性圖譜,得到濃度與溫度的標準曲線,如圖2所示,從圖2可得知:標準圖譜線性良好,相關係數R達到0.9959,最低檢測限可達到63pM。
2)過氧化氫(H2O2)水溶液濃度的檢測:將200μL的1nM的過氧化氫(H2O2)水溶液與800μL 200μg/mL吲哚菁綠修飾的NaLuF4:Yb,Er水溶液均勻混合,室溫(25℃)靜置反應10min後,反應已完全,在785nm近紅外光照射下測定混合液照射2min後的溫度變化,代入標準曲線,得到濃度為0.96nM。
實施例2、檢測過氧化氫(H2O2)水溶液的濃度:
1)標準曲線的繪製:將200μL的1nM、2nM、4nM,8nM、16nM的過氧化氫(H2O2)水溶液與800μL 200μg/mL銀包覆的NaGdF4:Yb,Er水溶液均勻混合,室溫(25℃)靜置反應20min後,反應已完全,在808nm近紅外光照射下測定混合液照射2min後的溫度變化,處理數據得到溫度的線性圖譜,得到濃度與溫度的標準曲線,如圖3所示,從圖3可得知:標準圖譜線性良好,相關係數R達到0.99678,最低檢測限可達到200pM。
2)過氧化氫(H2O2)水溶液濃度的檢測:將150μL的5nM的過氧化氫(H2O2)水溶液與800μL 200μg/mL銀包覆的NaGdF4:Yb,Er水溶液均勻混合,室溫(25℃)靜置反應20min後,反應已完全,在808nm近紅外光照射下測定混合液照射2min後的溫度變化,代入標準曲線,得到濃度為4.88nM。
實施例3、檢測抗壞血酸(AA)水溶液的濃度:
1)標準曲線的繪製:將200μL的500pM、1nM、2nM、4nM,8nM、16nM的抗壞血酸(AA)水溶液與800μL 200μg/mL三角形銀納米片水溶液均勻混合,室溫(25℃)靜置反應10min後,反應已完全,在980nm近紅外光照射下測定混合液照射2min後的溫度變化,處理數據得到溫度的線性圖譜,得到濃度與溫度的標準曲線,如圖6所示,從圖6可得知:標準圖譜線性良好,相關係數R達到0.99832,最低檢測限可達到50nM。
2)抗壞血酸(AA)水溶液濃度的檢測:將200μL的3nM的抗壞血酸(AA)水溶液與800μL 200μg/mL三角形銀納米片水溶液均勻混合,室溫(25℃)靜置反應10min後,反應已完全,在980nm近紅外光照射下測定混合液照射2min後的溫度變化,代入標準曲線,得到濃度為3.12nM。