2‑(2,3‑環氧丙基)苯酚組合物及製備方法與流程
2023-04-23 22:11:32 3
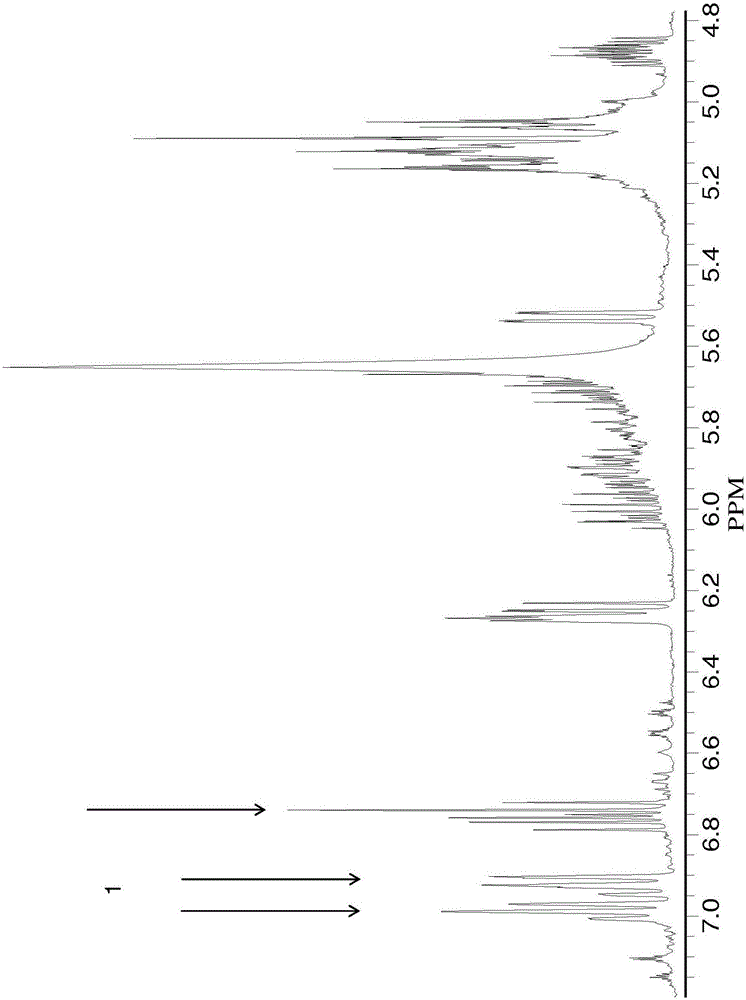
2-(2,3-環氧丙基)苯酚是其中2,3-環氧丙基或縮水甘油基直接鍵合至苯環碳原子的C-縮水甘油基苯酚,並且不同於其中縮水甘油基鍵合至苯酚氧原子的O-縮水甘油基苯酚或縮水甘油基苯基醚。由於2-(2,3-環氧丙基)苯酚的環氧官能度,使得其是熱固性樹脂中,特別是環氧樹脂中的潛在有用的反應物。為了成為商業材料,需要製備2-(2,3-環氧丙基)苯酚的有效、低成本方法。為了成本低,所述方法應該具有以下特性。它應該基於大批量可獲得的低成本材料,它應該是一步反應,並且它應該具有商業上可接受的循環時間(分批製備中各批次之間的時間)。技術實現要素:一種製備2-(2,3-環氧丙基)苯酚的方法包括使2-烯丙基苯酚與氧化劑在催化劑存在下反應,其中2-烯丙基苯酚包括並且2-(2,3-環氧丙基)苯酚包括其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基;或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基;並且其中所有重量百分數基於2-(2,3-環氧丙基)苯酚、2-烯丙基苯酚和3-色原烷醇的總重量。在另一個實施方式中,製備2-(2,3-環氧丙基)-6-甲基苯酚的方法包括使2-烯丙基-6-甲基苯酚與包括間氯過苯甲酸的氧化劑在包括雙(乙醯丙酮)二氧化鉬(VI)(bis(acetylacetonato)dioxomolybdenum(VI))、鎢酸、六羰基鎢或其組合的催化劑存在下反應。在另一個實施方式中,基於組合物的總重量,組合物包含:1至90重量百分數的以下結構的2-(2,3-環氧丙基)苯酚5至90重量百分數的以下結構的2-烯丙基苯酚和0至40重量百分數的以下結構的3-色原烷醇其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基;其中所有重量百分數基於2-(2,3-環氧丙基)苯酚、2-烯丙基苯酚和3-色原烷醇的總重量。另一實施方式是通過使以下結構的2-烯丙基苯酚與氧化劑在催化劑存在下反應製備的熱固性聚合物其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基。附圖說明現在參考附圖:圖1描繪了來自實施例4的反應混合物的400Mhz1H-NMR譜,所述反應混合物含有甲醇、未反應的2-烯丙基-6-甲基苯酚、8-甲基-3-色原烷醇和過氧化氫。圖2描繪了實施例3的反應混合物的400Mhz1H-NMR譜和2D-相關性(COSY)譜,所述反應混合物是2-烯丙基-6-甲基苯酚與間氯過苯甲酸在MoO2(acac)2存在下在30℃下反應5小時後的產物。圖3描繪了實施例3的反應混合物的8.2-6.8ppm範圍內的400Mhz1H-NMR譜。圖4描繪了實施例3的反應產物的6-2ppm範圍內的400Mhz1H-NMR譜。圖5描繪了實施例15的2-烯丙基-6-甲基苯酚與間氯過苯甲酸的MoO2(acac)2催化的環氧化中作為反應時間的函數繪製的2-烯丙基-6-甲基苯酚的轉化率,以及以重量百分數表示的2-(2,3-環氧丙基)-6-甲基苯酚和8-甲基-3-色原烷醇的量。圖6描繪了實施例16的2-烯丙基-6-甲基苯酚與間氯過苯甲酸的MoO2Cl2催化的環氧化中作為反應時間的函數繪製的2-烯丙基-6-甲基苯酚的轉化率,以及以重量百分數表示的2-(2,3-環氧丙基)-6-甲基苯酚和8-甲基-3-色原烷醇的量。圖7描繪了實施例17的2-烯丙基-6-甲基苯酚與間氯過苯甲酸的H2WO4催化的環氧化中作為反應時間的函數繪製的2-烯丙基-6-甲基苯酚的轉化率,以及以重量百分數表示的2-(2,3-環氧丙基)-6-甲基苯酚和8-甲基-3-色原烷醇的量。圖8描繪了實施例18的2-烯丙基-6-甲基苯酚與間氯過苯甲酸的W(CO)6催化的環氧化中作為反應時間的函數繪製的2-烯丙基-6-甲基苯酚的轉化率,以及2-(2,3-環氧丙基)-6-甲基苯酚和8-甲基-3-色原烷醇的量。具體實施方式為了成為商業材料,需要製備2-(2,3-環氧丙基)苯酚的有效、低成本方法。本發明人已經確定了用於製備2-(2,3-環氧丙基)苯酚的直接一步路徑,其是相應2-烯丙基苯酚的環氧化。2-烯丙基苯酚是其中烯丙基直接鍵合至苯基碳原子的烯丙基苯酚,並且不同於其中烯丙基直接鍵合至苯酚的氧的O-烯丙基苯酚。所述方法適用於2-烯丙基苯酚,如2-烯丙基-6-甲基苯酚,並可以利用市售的過氧化物,例如,間氯過苯甲酸。所述方法具有一個反應步驟,即2-烯丙基苯酚的環氧化。此外,2-烯丙基苯酚的轉化可以快速進行,這提供了商業上可接受的循環時間。因此,製備2-(2,3-環氧丙基)苯酚的方法包括使2-烯丙基苯酚與氧化劑在催化劑存在下進行反應;其中2-烯丙基苯酚包括:並且2-(2,3-環氧丙基)苯酚包括:其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基;並且其中所有重量百分數基於2-(2,3-環氧丙基)苯酚、2-烯丙基苯酚和3-色原烷醇的總重量。如在本文所用的,術語「烴基」廣泛地指包含碳和氫,可選地具有1至3個雜原子,例如,氧、氮、滷素、矽、硫或其組合的單價取代基。在一些實施方式中,Z基團選自氫、甲基及其組合。例如,所述方法可以用於由2-烯丙基-6-甲基苯酚製備2-(2,3-環氧丙基)-6-甲基苯酚。催化劑可以是過渡金屬催化劑。例如,催化劑可以是包含鉬、釩、鎢、鈦、錳、鈮或其組合的過渡金屬催化劑。過渡金屬可以處於IV、V或VI氧化態,例如,Ti(IV)、Nb(V)、Mn(VI)、Mo(VI)或W(VI)。具體的催化劑包括但不限於,雙(乙醯丙酮)二氧化鉬(VI)(MoO2(acac)2)、二氯二氧化鉬(molybdenumdichloridedioxide)(MoO2Cl2)、鎢酸(H2WO4)、鎢矽酸(H4O40SiW12·xH2O)、六羰基鎢(W(CO)6)、六羰基鉬(Mo(CO)6)、乙醯丙酮釩(V(acac)2)、乙醯丙酮氧釩(VO(acac)2)、五氧化二釩(V2O5)、或其組合。在一些實施方式中,催化劑包括雙(乙醯丙酮)二氧化鉬(VI)、二氯二氧化鉬、鎢酸、六羰基鎢、或其組合。催化劑也可以是在惰性固體載體例如二氧化矽或氧化鋁上的非均相催化劑。氧化劑可以包括有機過氧化物,即含有過氧「-O-O-」基團的有機化合物。具體而言,氧化劑可以包括過氧化氫、烷基過氧化物、烷基氫過氧化物(alkylhydroperoxide)、酮過氧化物(ketoneperoxide)、二醯基過氧化物、二過氧縮酮(diperoxyketal)、過氧化酯、過氧二碳酸酯、過氧酸、過苯甲酸、或其組合。氧化劑的實例包括過氧化氫、2-丁酮過氧化物、環己酮過氧化物、過氧化苯甲醯、月桂基過氧化物、二叔丁基過氧化物、叔丁基枯基過氧化物、二枯基過氧化物、叔丁基氫過氧化物、氫過氧化枯烯、過氧苯甲酸叔丁酯、過氧苯甲酸叔戊酯、過氧辛酸叔丁酯、2,2-二(叔丁基過氧)丁烷、2,2-二(叔丁基過氧)辛烷、2,5-二甲基己烷-2,5-二氫過氧化物、2,5-二甲基-2,5-二(苯甲醯基過氧)己烷、2,5-二甲基-2,5-二(叔丁基過氧)己烷、2,5-二甲基-2,5-二(叔丁基過氧)己-3-炔、1,4-二(叔丁基過氧異丙基)苯、1,3-二(叔丁基過氧異丙基)苯、二(叔丁基過氧)間苯二甲酸酯、過甲酸、過乙酸、過丙酸、過丁酸、過異戊酸、過庚酸、過苯甲酸、間氯過苯甲酸、單過鄰苯二甲酸、對甲氧基過苯甲酸、間硝基過苯甲酸、α-過萘甲酸、β-過萘甲酸、二(三甲基甲矽烷基)過氧化物、三甲基甲矽烷基三苯基甲矽烷基過氧化物、及其組合。在一些實施方式中,氧化劑包括間氯過苯甲酸(MCPBA),並且在一些實施方式中,氧化劑包括過氧化氫。所述方法可以可選地在溶劑的存在下進行。如實施例1中的記錄,在選擇溶劑時可能要考慮的實際因素包括成本、健康危害性、2-烯丙基苯酚的溶解度、氧化劑、催化劑的溶解度、以及沸點。例如,溶劑可以是非極性溶劑,例如,苯、甲苯、乙苯、枯烯、二甲苯、均三甲苯、四氫化萘、氯苯、二氯苯、氯仿、或其組合。溶劑也可以是極性溶劑,例如,水、甲醇、乙醇、2-丙醇、丙酮、甲乙酮、二甲氧基乙烷、四氫呋喃、1,4-二噁烷、二甲基甲醯胺、二甲基亞碸、乙腈、或其組合。也可以使用前述溶劑的任何組合,包括極性和非極性溶劑的組合。甲醇或2-丙醇由於易於通過蒸餾從產物中除去,與含水過氧化氫的混溶性和低成本而是有利的。因此,在一些實施方式中,溶劑包括甲醇、2-丙醇或其組合。溶劑也可以是水。甲苯和氯仿由於是2-烯丙基苯酚和2-(2,3-環氧丙基)苯酚的良好溶劑並是催化劑的不良溶劑而因此簡化催化劑去除和產物分離,是有利的。因此,在一些實施方式中,溶劑包括甲苯、氯仿、或其組合。環氧化可以在寬範圍的溫度和時間內進行,這部分取決於所用的具體氧化劑和催化劑。如本文所用,術語「環氧化」和「反應」均指在氧化劑和催化劑存在下由2-烯丙基苯酚製備2-(2,3-環氧丙基)苯酚的本發明方法。反應溫度是反應時間和氧化劑分解速率之間的平衡。如果溫度太低,反應將需要很長時間。如果反應溫度太高,氧化劑將在其有機會與2-烯丙基苯酚反應之前就會發生分解。為了實現經濟上可行的反應時間,最低反應溫度可以是-20、0或20℃。最高溫度部分地由氧化劑的分解溫度確定。例如,間氯過苯甲酸在約80℃開始分解。對於一些氧化劑,包括間氯過苯甲酸,反應溫度可以是-20至80℃,具體是0至60℃,且更具體是20至40℃。過氧化氫在約40℃開始分解。因此,當使用過氧化氫作為氧化劑時,反應溫度可以為-20至50℃,具體是0至45℃,且更具體是20至40℃。反應時間可以為10分鐘至8小時,具體為20分鐘至6小時,且更具體是30分鐘至4小時。在一些實施方式中,反應溫度為-10至50℃,且反應時間為10分鐘至6小時。在任何反應溫度下,轉化率取決於氧化劑、催化劑的活性和反應時間。例如,採用作為氧化劑的間氯過苯甲酸和鉬催化劑如MoO2(acac)2和MoO2Cl2,在前十分鐘內發生50%的2-烯丙基苯酚轉化(圖5和6)。採用作為氧化劑的間氯過苯甲酸和鎢催化劑如H2WO4和W(CO)6,在前30分鐘內發生60%的2-烯丙基苯酚轉化,另外20%的轉化發生於70分鐘以上的時間內(圖7和圖8)。在一些實施方式中,在-10至50℃的反應溫度下,在10分鐘至6小時內存在2-烯丙基苯酚的50至100%轉化。在一些實施方式中,2-烯丙基苯酚包括2-烯丙基-6-甲基苯酚且2-(2,3-環氧丙基)苯酚包括2-(2,3-環氧丙基)-6-甲基苯酚。製備2-(2,3-環氧丙基)-6-甲基苯酚的方法包括使2-烯丙基-6-甲基苯酚與包括間氯過苯甲酸的氧化劑在催化劑存在下反應,所述催化劑包括雙(乙醯丙酮)二氧化鉬(VI)、鎢酸、六羰基鎢、或其組合。在環氧化反應中,可以形成以下結構的3-色原烷醇其中每個Z獨立地如對於2-烯丙基苯酚和2-(2,3-環氧丙基)苯酚定義的。因此,可以形成2-(2,3-環氧丙基)苯酚和3-色原烷醇的混合物。在一些實施方式中,基於組合物的總重量,組合物包含0至40重量百分數,具體1至40重量百分數,且更具體1至20重量百分數的3-色原烷醇。此外,如果2-烯丙基苯酚的轉化不完全,可以形成2-烯丙基苯酚、2-(2,3-環氧丙基)苯酚和3-色原烷醇的混合物。因此,在一些實施方式中,2-烯丙基苯酚與氧化劑在催化劑存在下的反應可以產生包含下述的組合物,基於組合物的總量,1至90重量百分數的以下結構的2-(2,3-環氧丙基)苯酚5至90重量百分數的以下結構的2-烯丙基苯酚和0至40重量百分數的以下結構的3-色原烷醇其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基;並且其中所有重量百分數基於2-(2,3-環氧丙基)苯酚、2-烯丙基苯酚和3-色原烷醇的總重量。在一些實施方式中,2-(2,3-環氧丙基)苯酚包括2-(2,3-環氧丙基)-6-甲基苯酚,2-烯丙基苯酚包括2-烯丙基-6-甲基苯酚,且3-色原烷醇包括其在本文中稱為「8-甲基-3-色原烷醇」或「MCO」。基於組合物的總重量,組合物可以包含0至40重量百分數,具體1至40重量百分數,且更具體1至20重量百分數的8-甲基-3-色原烷醇。因此,在一些實施方式中,基於組合物的總重量,組合物包含1至90重量百分數的2-(2,3-環氧丙基)-6-甲基苯酚、5至90重量百分數的2-烯丙基-6-甲基苯酚、和0至40重量百分數的8-甲基-3-色原烷醇。取決於催化劑,產生的2-(2,3-環氧丙基)苯酚的量可能由於導致由2-烯丙基苯酚形成3-色原烷醇的次級環化反應而隨時間降低。例如,如由圖5可以看出,當2-烯丙基苯酚是2-烯丙基-6-甲基苯酚時,氧化劑是間氯過苯甲酸,並且催化劑是MoO2(acac)2,則在反應的前20分鐘獲得的2-(2,3-環氧丙基)-6-甲基苯酚的最大量為約35重量百分數。最終的2-(2,3-環氧丙基)-6-甲基苯酚的量為約30重量百分數。因此,在一些實施方式中,基於2-(2,3-環氧丙基)-6-甲基苯酚、2-烯丙基-6-甲基苯酚和8-甲基-3-色原烷醇的總重量,組合物包含1至40重量百分數的2-(2,3-環氧丙基)-6-甲基苯酚、30至90重量百分數的2-烯丙基-6-甲基苯酚以及1至40重量百分數的8-甲基-3-色原烷醇。如由圖6中可以看出,採用MoO2Cl2,在10min內獲得的2-(2,3-環氧丙基)-6-甲基苯酚的最大量為15重量百分數。在約22分鐘後,未觀察到2-(2,3-環氧丙基)-6-甲基苯酚,但形成了25重量百分數的8-甲基-3-色原烷醇。如由圖7中可以看出,採用H2WO4,在前20分鐘內獲得了65重量百分數的2-(2,3-環氧丙基)-6-甲基苯酚,且在另外80min之後獲得了85重量百分數的2-(2,3-環氧丙基)-6-甲基苯酚。由圖8可以看出,採用W(CO)6的結果與採用H2WO4的結果相似。因此,在一些實施方式中,基於組合物的總重量,基於2-(2,3-環氧丙基)-6-甲基苯酚、2-烯丙基-6-甲基苯酚和8-甲基-3-色原烷醇的總重量,組合物包含5至90重量百分數的2-(2,3-環氧丙基)-6-甲基苯酚、5至90重量百分數的2-烯丙基-6-甲基苯酚、和1至20重量百分數的8-甲基-3-色原烷醇。在一些條件下,2-烯丙基苯酚,例如,2-烯丙基-6-甲基苯酚,與氧化劑在催化劑存在下的反應可以導致熱固性聚合物的形成。熱固性聚合物可以具有230至260℃,具體230至250℃,且更具體235至245℃的玻璃化轉變溫度。具體而言,2-烯丙基-6-甲基苯酚與氧化劑在催化劑存在下的反應可以導致玻璃化轉變溫度為235至245℃的熱固性聚合物。例如,2-烯丙基-6-甲基苯酚與過氧化氫水溶液在MoO2(acac)2和甲醇存在下的反應,如實施例4中一樣,隨後在30至65℃下加熱,如實施例19中一樣,產生玻璃化轉變溫度為241℃的熱固性聚合物。本發明至少包括以下實施方式。實施方式1:一種製備2-(2,3-環氧丙基)苯酚的方法,包括在催化劑存在下使2-烯丙基苯酚與氧化劑反應;其中2-烯丙基苯酚包括並且2-(2,3-環氧丙基)苯酚包括其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基。實施方式2:根據實施方式1的方法,其中2-烯丙基苯酚包括2-烯丙基-6-甲基苯酚且2-(2,3-環氧丙基)苯酚包括2-(2,3-環氧丙基)-6-甲基苯酚。實施方式3:根據實施方式1或2的方法,其中催化劑是包含鉬、釩、鎢、鈦、錳、鈮或其組合的過渡金屬催化劑。實施方式4:根據實施方式1-3中任一項的方法,其中催化劑包含雙(乙醯丙酮)二氧化鉬(VI)、二氯二氧化鉬、鎢酸、六羰基鎢、或其組合。實施方式5:根據實施方式1-4中任一項的方法,其中氧化劑包括過氧化氫、烷基過氧化物、烷基氫過氧化物、酮過氧化物、二醯基過氧化物、二過氧縮酮、過氧化酯、過氧二碳酸酯、過氧酸、過苯甲酸或其組合。實施方式6:根據實施方式1-5中任一項的方法,其中氧化劑包括間氯過苯甲酸。實施方式7:根據實施方式1-6中任一項的方法,其中氧化劑包括過氧化氫。實施方式8:根據實施方式1-7中任一項的方法,其中反應溫度為-10至50℃,且反應時間為10分鐘至6小時。實施方式9:根據實施方式1-8中任一項的方法,其中在-10至50℃的反應溫度下,2-烯丙基苯酚在10分鐘至6小時內轉化了50至100%。實施方式10:根據實施方式1-9中任一項的方法,其中對於2-(2,3-環氧丙基)苯酚的選擇性為50至100%。實施方式11:根據實施方式1-10中任一項的方法,其中以下結構的3-色原烷醇與2-(2,3-環氧丙基)苯酚一起製備,其中Z如在實施方式1中定義的。實施方式12:根據實施方式1-11中任一項的方法,其中:2-烯丙基苯酚包括2-烯丙基-6-甲基苯酚;2-(2,3-環氧丙基)苯酚包括2-(2,3-環氧丙基)-6-甲基苯酚;氧化劑包括間氯過苯甲酸;並且催化劑包括雙(乙醯丙酮)二氧化鉬(VI)、鎢酸、六羰基鎢或其組合。實施方式12a:一種製備2-(2,3-環氧丙基)-6-甲基苯酚的方法,包括使2-烯丙基-6-甲基苯酚與包括間氯過苯甲酸的氧化劑在包含雙(乙醯丙酮)二氧化鉬(VI)、鎢酸、六羰基鎢或其組合的催化劑存在下反應。實施方式13:一種組合物,包含:1至90重量百分數的以下結構的2-(2,3-環氧丙基)苯酚5至90重量百分數的以下結構的2-烯丙基苯酚和0至40重量百分數的以下結構的3-色原烷醇其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基,並且其中所有重量百分數基於2-(2,3-環氧丙基)苯酚、2-烯丙基苯酚和3-色原烷醇的總重量。實施方式14:根據實施方式13的組合物,其中2-(2,3-環氧丙基)苯酚包括2-(2,3-環氧丙基)-6-甲基苯酚,2-烯丙基苯酚包括2-烯丙基-6-甲基苯酚,並且3-色原烷醇包括8-甲基-3-色原烷醇。實施方式15:根據實施方式13或14的組合物,基於2-(2,3-環氧丙基)-6-甲基苯酚、2-烯丙基-6-甲基苯酚和8-甲基-3-色原烷醇的總重量,包含1至40重量百分數的2-(2,3-環氧丙基)-6-甲基苯酚、30至90重量百分數的2-烯丙基-6-甲基苯酚和1至40重量百分數的8-甲基-3-色原烷醇。實施方式16:根據實施方式13的組合物,基於2-(2,3-環氧丙基)-6-甲基苯酚、2-烯丙基-6-甲基苯酚和8-甲基-3-色原烷醇的總重量,包含5至90重量百分數的2-(2,3-環氧丙基)-6-甲基苯酚、5至90重量百分數的2-烯丙基-6-甲基苯酚和1至20重量百分數的8-甲基-3-色原烷醇。實施方式17:一種通過使以下結構的2-烯丙基苯酚與氧化劑在催化劑存在下反應製備的熱固性聚合物其中每次出現的Z獨立地為氫;滷素;未取代或取代的C1-C12烴基,條件是烴基不是叔烴基;C1-C12烴硫基;C1-C12烴氧基或其中至少兩個碳原子將滷素和氧原子分開的C2-C12滷代烴氧基。實施方式18:根據實施方式17的熱固性聚合物,其中2-烯丙基苯酚包括2-烯丙基-6-甲基苯酚,並且熱固性聚合物具有235至245℃的玻璃化轉變溫度。本發明通過以下非限制性實施例進一步舉例說明。實施例實施例中使用的材料列於下表1中。表1.材料CAS號化學名純度(wt%)縮寫3354-58-32-烯丙基-6-甲基苯酚50%AMP1567906-11-92-(2,3-環氧丙基)-6-甲基苯酚可變EMP136514-01-78-甲基-3-色原烷醇可變MCO7722-84-1水性H2O2/H2O50%-75-05-8乙腈>99.9%-67-66-3氯仿>99.9%-865-49-6氯仿-d99.8%CDCl368-12-2N,N-二甲基甲醯胺>99.9%DMF123-91-11,4-二噁烷>99%-67-56-1甲醇99%-78-93-3甲基乙基酮>99%MEK109-99-9四氫呋喃>99.9%THF108-88-3甲苯99%-937-14-4間氯過苯甲酸77%MCPBA63393-96-4三辛基單甲基氯化銨99%ALIQUATTM336139-13-92,2',2"-次氮基三乙酸-NTA13939-06-5六羰基鉬99.9%-17524-05-9雙(乙醯丙酮)二氧化鉬(VI)-MoO2(acac)213637-68-8二氯二氧化鉬-MoO2Cl214040-11-0鎢酸水合物97%H2WO47783-03-1六羰基鎢99%W(CO)612027-43-9鎢矽酸99.9%H4O40SiW12.xH2O13476-99-8乙醯丙酮釩97%V(acac)23153-26-2乙醯丙酮氧釩98%VO(acac)21314-62-1五氧化二釩-V2O5通過1H-NMR光譜表徵反應混合物。反應混合物的1H-NMR譜在圖1至4中描述。在圖2至4中,1H-NMR峰通過對應於所示的其中化學結構中的具體氫原子的數目標識。實施例1.溶劑篩選對50wt%H2O2/H2O和AMP在各種溶劑中的溶解度的觀察結果總結於表2中。所有溶解度的研究在室溫和大氣壓下進行。將氧化劑(水性H2O2)和溶質(AMP)與表2中所列的溶劑混合,並記錄觀察結果。發現H2O2和AMP在極性溶劑中完全混溶。AMP可溶於芳族溶劑中,而水性H2O2不溶,導致形成兩相。為了比較溶劑,成本、健康危害、H2O2和AMP溶解度、催化劑溶解度和基於沸點的溶劑分離容易度被賦予正(+)或負(-)權重。結果總結於表3中。這種操練將甲醇定為潛在溶劑。與所評價的其它溶劑相比,甲醇相對便宜且無危險。此外,甲醇的低沸點使其分離和回收比其他溶劑能量密集更低。據發現,除鎢酸外的所有候選催化劑都不溶於甲醇,因此可以通過過濾回收。鎢酸微溶於甲醇。表2.50%H2O2/H2O和2-烯丙基-6-甲基苯酚在各種溶劑中的溶解度a.由移液管溶解的聚(丙烯腈-丁二烯-苯乙烯)。表3.溶劑選擇採納的標準a.由移液管溶解的聚(丙烯腈-丁二烯-苯乙烯)。b.成本估算是定性的並基於少量購買的CHROMASOLVTM級溶劑。定價信息獲自Sigma-Aldrich。價格高於US$70/升被認為是不合需要的。c.2或更大的HMIS健康危害認為是不合需要的。d.「+」表示催化劑不溶。催化劑形成非均相反應混合物的不溶性消除了催化劑分離問題。實施例2.水性H2O2和2-烯丙基-6-甲基苯酚的MoO2(acac)2-2催化環氧化反應混合物(AMP+H2O2+CH3OH+MoO2(acac)2)最初是無色的,並在反應的前半小時內變成深棕色。這種顏色變化可以歸因於鉬二過氧物種的形成(導致鉬氧化態的變化)。獲得的結果強烈依賴於反應溫度。在15-20℃的低反應溫度下,發現AMP的信號強度(通過GC/MS測定,通過GC測定的AMP的峰高)對於初始和最終樣品是相似的,表明在這些溫度下沒有反應。另一方面,在40℃的較高溫度下觀察到AMP峰的峰面積顯著降低(用GC/MS和1H-NMR確認)。理論上估計的和通過反應混合物的GC/MS分析獲得的產物離子的質量與EMP的形成一致。EMP外標的不可用性和在儀器資料庫和開放文獻中對於EMP的模型MS質譜不存在,都降低了MS軟體預測的化學結構的置信度。此外,環氧化AMP(EMP)和環化的AMP(8-甲基-3-色原烷醇)的相同精確質量消除了MS區分這些反應產物的使用。圖1描述了實施例4的反應混合物的1H-NMR光譜,其含有未反應的AMP、甲醇、8-甲基-3-色原烷醇。如由圖1可以看出,與未反應的AMP的1H-NMR光譜中的相同峰相比,圖中「1」表示的7ppm(雙峰)和6.7ppm(三峰)處的吸收峰強度的降低表明AMP的碳-碳雙鍵的反應已經發生。與來自甲醇的超強吸收峰耦合的繁忙的(被佔用的,busy)1H-NMR譜阻礙了通過1H-NMR鑑定EMP。樣品需要在分析前通過除去甲醇和殘留的過氧化氫進行清潔。當將W類和其它Mo類催化劑用於環氧化反應時,獲得類似的結果,對應於碳-碳雙鍵和其它峰形成的吸收峰強度的降低。因此,已經證明AMP的碳-碳雙鍵的反應。實施例3-14.2-烯丙基-6-甲基苯酚的金屬催化環氧化與實施例2中的反應產物的鑑定相關的問題通過代替甲醇在CDCl3中進行AMP環氧化反應而消除。使用CDCl3消除了對溶劑分離的需要,從而能夠通過NMR進行產物的確定性鑑定,而不需要加熱或以其它方式濃縮樣品。由於H2O2與氯仿的不混溶性,MCPBA用作H2O2的替代物。通過代替甲醇或氯仿在CDCl3中進行催化劑篩選研究,也使樣品製備所需的時間最小化。研究了AMP使用MCPBA或H2O2的金屬催化環氧化以確定用於選擇性形成EMP的最佳催化劑。實施例3-13的反應物和結果總結於表4中。大多數研究的催化劑使用以下反應物進行:AMP+MCPBA+催化劑。在實施例4、9和11中,用H2O2代替MCPBA。溶劑是實施例1、3-10和12-14中的CDCl3。在實施例4和11中,用甲醇代替CDCl3。反應混合物的初始組成為5.02×10-3molAMP+8.6×10-3mol氧化劑+0.18mol溶劑。所用的氧化劑是CDCl3中的MCPBA和甲醇或CDCl3中的50wt%H2O2/H2O。反應在25℃進行6小時。在AMP與MCPBA或50wt%H2O2/H2O的反應中AMP(柱3)的轉化率和EMP(柱4)和8-甲基-3-色原烷醇(柱5)的量和選擇性和各種金屬催化劑總結於表4中。在實施例4、9和11中觀察到溫度放熱,其中氧化劑為50wt%H2O2/H2O。當釩催化劑與50wt%H2O2/H2O組合使用時,當將催化劑引入反應混合物中時,觀察到H2O2高度放熱分解。即使在非常低的釩催化劑負載下也觀察到放熱氫H2O2分解。MCPBA可溶於CDCl3中,而反應的副產物,即在氧原子轉移到AMP後形成的間氯過苯甲酸,是不溶的。因此,在與MCPBA的環氧化中形成不溶性固體用作反應的間接指示。在催化劑存在下不存在MCPBA分解,在單獨的測試中都得到證實。在CDCl3中的金屬催化劑存在下AMP與MCPBA環氧化之後獲得的產物混合物的1H-NMR譜與在甲醇中獲得的反應產物的NMR譜相比相對乾淨。實施例5和8中EMP的形成通過1H-NMR譜證實。反應混合物還通過2D相關光譜(COSY)進行研究。反應混合物的示例性COSY光譜在圖2中再現。從COSY譜中鑑定出8-甲基-3-色原烷醇的存在。8-甲基-3-色原烷醇的形成是分子內環化反應,並可以歸因於苯酚羥基和EMP的環氧化物基團之間的反應。藉助COSY光譜鑑定反應產物的1H-NMR譜中對應於8-甲基-3-色原烷醇的那些吸收峰。一旦鑑定出8-甲基-3-色原烷醇峰,就使用標準1H-NMR譜對表4中總結的催化劑篩選研究中反應產物中的8-甲基-3-色原烷醇的量進行定量。存在於AMP的烯丙基側鏈中的碳-碳雙鍵經歷環氧化,導致EMP的形成。8-甲基-3-色原烷醇可以通過EMP的分子內環化形成。因此,8-甲基-3-色原烷醇的形成表明至少作為中間體的EMP的形成。通過1H-NMR譜測定的對8-甲基-3-色原烷醇幾乎完全的選擇性表明長反應時間有利於EMP的環化。除了長反應時間,8-甲基-3-色原烷醇的容易形成也可以歸因於環氧化物基團的高反應性和對色原烷環形成的空間位阻的缺乏。實施例3.MCPBA和MoO2(acac)2催化劑將0.75mLAMP、1.5gMCPBA、400mgMoO2(acac)2和CDCl3進料到容器中。將容器置於冰浴中進行冷卻。將混合物用磁力攪拌棒進行攪拌。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例4.水性H2O2和MoO2(acac)2催化劑將3mLAMP、4mL50wt%H2O2/H2O、400mgMoO2(acac)2和甲醇進料到容器中。反應在接近環境條件下進行,並將混合物用磁力攪拌棒攪拌。在25℃下攪拌6小時後,通過GC-MS分析反應混合物。實施例5.MCPBA和MoO2Cl2催化劑將0.75mLAMP、1.5gMCPBA、400mgMoO2Cl2和CDCl3加入容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例6.MCPBA和Mo(CO)6催化劑將0.75mLAMP、1.5gMCPBA、400mgMo(CO)6和CDCl3進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例7.MCPBA和H2WO4催化劑將0.75mLAMP、1.5gMCPBA、200mgH2WO4和CDCl3進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例8.MCPBA和W(CO)6催化劑將0.75mLAMP、1.5gMCPBA、200mgW(CO)6和CDCl3進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例9.水性H2O2和W(CO)6催化劑將0.75mLAMP、4mL50wt%H2O2/H2O、200mgW(CO)6和CDCl3進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例10.MCPBA和H4O40SiW12·xH2O催化劑將0.75mLAMP、1.5gMCPBA、200mgH4O40SiW12.xH2O和CDCl3進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例11.水性H2O2和H4O40SiW12·xH2O催化劑將0.75mLAMP、4mL50wt%H2O2/H2O、200mgH4O40SiW12.xH2O和甲醇進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例12.MCPBA和V(acac)2催化劑將0.75mLAMP、1.5gMCPBA、250mgV(acac)2和CDCl3進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例13.MCPBA和VO(acac)2催化劑將0.75mLAMP、4mL50wt%H2O2/H2O、250mgVO(acac)2和CDCl3進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。實施例14.MCPBA和V2O5催化劑將0.75mLAMP、1.5gMCPBA和250mgV2O5進料到容器中。通過將容器置於冰浴中冷卻容器,同時用磁力攪拌棒攪拌混合物。在25℃下攪拌6小時後,通過1H-NMR分析反應混合物。表4.AMP金屬催化環氧化中AMP的轉化率,EMP和MCO的收率a)除了MCO之外,還形成了許多未完全表徵的副產物。b)括號中是EMP或MCO基於反應的AMP的百分比選擇性。當使用MoO2(acac)2作為環氧化催化劑(實施例3和4)時,沒有檢測到EMP,這表明任何形成的EMP都環化成8-甲基-3-色原烷醇。對於W(CO)6催化反應的AMP的轉化率用MCPBA(實施例8)比用H2O2(實施例9)高得多。在W(CO)6催化(實施例8)和MoO2Cl2催化(實施例5)反應中採用MCPBA作為氧化劑觀察到EMP。AMP在實施例9中未反應可能是由於兩相H2O2/H2O+CDCl3反應混合物的兩個相之間受限的質量傳遞所致。H2O2存在於含水相中,而AMP存在於有機相(CDCl3)中。在所有測試的催化劑中,對於其優異的催化活性選擇四種催化劑MoO2(acac)2、MoO2Cl2、W(CO)6和H2WO4。催化劑研究以規則的時間間隔取樣進行,以允許優化反應時間、控制EMP收率的參數。實施例15-18.2-烯丙基-6-甲基苯酚的金屬催化環氧化的動力學研究AMP、EMP和8-甲基-3-色原烷醇的量作為實施例15-18中AMP的金屬催化環氧化的反應時間的函數分別繪製於圖5-8中。在圖中,AMP的轉化%由實心菱形表示;EMT量由實心方塊表示;且8-甲基-3-色原烷醇量由實心三角形表示。對於每個實施例,氧化劑是MCPBA,且溶劑是CDCl3。在實施例15和圖5中,在MoO2Cl2存在下AMP的轉化率為約60%。在前20分鐘內消耗了這個量的AMP約90%。EMP和8-甲基-3-色原烷醇的量都為約30%。相比之下,實施例16和圖6中,在MoO2(acac)2存在下的AMP轉化率僅為約25%,且在前20分鐘後沒有觀察到EMP。與Mo類催化劑相比,使用W類催化劑觀察到EMP更高的選擇性。在實施例17和圖7中,在鎢酸存在下AMP的轉化率為約90%。EMP和8-甲基-3-色原烷醇的量分別為約80%和約10%。在實施例18和圖8中,採用W(CO)6的AMP轉化率為約80%。在前30分鐘內,這個量的AMP約80%發生轉化。EMP和8-甲基-3-色原烷醇的量分別為約70%和約10%。如圖15-18中可以看出,8-甲基-3-色原烷醇在反應早期形成。因此,優化(最小化)反應時間不會消除8-甲基-3-色原烷醇的形成。實施例19.衍生自2-烯丙基-6-甲基苯酚的熱固性聚合物首先在氮氣覆蓋層下在約60-70℃下從實施例4的反應產物中蒸發甲醇後,通過1H-NMR譜法直接證實EMP形成的努力失敗了。除去溶劑導致在氯仿和DMSO中都不溶的粉末形成,表明形成了交聯的聚合物。使用DSC的熱分析鑑定不溶性粉末是具有241℃的玻璃化轉變溫度(Tg)的熱固性聚合物。用TGA對這種物質的進一步熱表徵表明在空氣中在600、700和800℃下焦炭質量分別為16.47%、16.60%和16.58%,且在氮氣氛下分析時焦炭質量分別為75.64%(600℃)、70.81%(700℃)和62.90%(800℃)。如本文中所用,術語「烴基」和「烴」泛指包含碳和氫,可選地具有1至3個雜原子,例如,氧、氮、滷素、矽、硫或其組合的取代基。在描述本發明的上下文中(特別是在所附權利要求的上下文中)使用術語「一個」、「一種」、「該」和類似指示物,除非本文另有說明或與上下文明顯矛盾,應該解釋為涵蓋單數和複數。「或」是指「和/或」。涉及相同組分或性質的所有範圍的端點都包括性的並可獨立地組合。除了更寬的範圍或更大的基團之外,更窄範圍或更具體的基團的公開並非是對更寬範圍或更大基團的免責聲明。本文公開的所有範圍包括端點,並且端點可彼此獨立地組合。如本文所用的術語「第一」和「第二」等不表示任何順序、數量或重要性,而是僅用於表示一個要素有別於另一個。本文所用的「包含」包括實施方式「基本上由列出的要素組成」或「由其組成」。除非另有定義,本文使用的技術和科學術語具有與本發明所屬領域技術人員通常理解的相同含義。「組合」包括共混物、混合物、反應產物等。雖然為了舉例說明的目的已經闡述了典型的實施方式,但是前面的描述不應當認為是對本文範圍的限制。因此,在不背離本文的精神和範圍的情況下,本領域技術人員能夠想到各種修改、改編和替代。當前第1頁1 2 3