新的光致變色化合物和用於生產光致變色化合物的中間化合物的製作方法
2023-05-14 04:44:37 2
本發明涉及新的雙環橋接[1,3]氧氮雜卓衍生物,其具備增強的光致變色特性且可用作分子光開關。
本發明還公開生產所述新的光致變色化合物的方法以及用於製備該化合物的中間物。
背景技術:
已知各類光致變色化合物:吲哚啉螺吡喃、吲哚啉螺惡嗪、苯並吡喃和萘並吡喃、俘精酸酐、二芳基乙烯、醌、呸啶螺環己二烯酮[v.malatesta,photodegradationoforganicphotochromesinorganicphotochromicandthermochromiccompounds.第2卷.j.c.crano及r.j.guglielmetti編.kluweracademic/plenumpublishers,newyork,1991,第473頁]、吲哚並[2,1-b][1,3]苯並惡嗪。使螺吡喃和螺惡嗪溶液暴露於uv輻射阻斷c-o鍵且形成著色的部花青,其由於分子的熱運動或在微秒及毫秒內受可見光輻射後而恢復至初始狀態。已知的光致變色化合物–吲哚啉並螺吡喃例如在專利ep0411884a1、gb1270928a、jp2006249622a、us5155230、us5241075及其他專利中描述。二氫吲哚啉與吲哚啉並螺吡喃類似,在受uv輻射後轉變成著色形式且保溫後恢復至初始狀態,但著色形式的壽命與吲哚啉螺吡喃相比更長,且長達數百毫秒、分鐘或甚至小時。[wo2012172093;r.zemribo、j.fotins、u.a.holgerkubas等人,photochromismofdihydroindolizines:第xiv部分。synthesisandphotophysicalbehaviorofphotochromicdihydroindolizinetripodallinkerstowardanchoringsensitizerstosemiconductornanoparticles.j.phys.org.chem.2011,24173-184;t.b.shrestha、j.melin、y.liu等人,newinsightsinthephotochromicspiro-dihydroindolizine/betaine-system.photochem.photobiol.sci.,2008,7,1449-1456]。已知的另一類光致變色化合物–醌類也未展示非著色形式與著色形式之間的極快互換,其著色轉換耗時數微秒至數百微秒。[n.p.gritsan、l.s.klimenko.j.photochem.photobiol.a:chem.1993,70,103-117]。
呸啶環己二烯酮的特徵在於著色形式的壽命極長,由於z-e構形交換而達到數天(與部花青中的e-z類似)。相對近期發現的光致變色化合物苯並吡喃和萘並吡喃不具備足夠的光穩定性。由於uv輻射的作用,吡喃環轉變成呋喃環且初始化合物喪失光致變色特性。[c.d.gabbutt、b.m.heron、s.b.kolla等人。ringcontractionduringthe6p-electrocyclisationofnaphthopyranvalencetautomers.orgbiomolchem2008;6:3096-3104;us6410754b1]。俘精酸酐和俘精醯亞胺的特徵在於在暴露於uv輻射時發生電環化反應,此時形成具有著色的1,3-環己二烯片段的化合物。後者熱穩定且只在受可見光輻射後可轉變成初始狀態。同樣熱穩定的二芳基乙烯的作用類似。
最新的一些種類的光致變色化合物-吲哚[2,1-b][1,3]苯並惡嗪具有熱不穩定的著色形式,但它們比吲哚啉螺吡喃快得多且其壽命比吲哚啉螺吡喃的壽命短[us8252209]。新的一類光致變色化合物6-硝基-1,3,3『,4-四氫螺[苯並吡喃-2,2『-吲哚]在lithuanian專利lt6024(wo2014/035244)中描述,其著色形式的壽命僅為22-41ns。
當吲哚啉螺吡喃暴露於紫外線輻射時,發生吡喃環破裂且出現著色的部花青形式[v.i.minkin,photoswitchablemolecularsystemsbasedonsipropyransandspirooxazines,第37-80頁.inmolecularswitches/由b.l.feringa和w.r.browne編.weinheim,wiley-vch,2011,第1-2卷;spiropyrans.r.c.bertelson,第11-84頁.inorganicphotochromicandthermochromiccompounds,第1-2卷,j.c.crano,r.j.guglielmetti編,plenumpress.newyorkandlondon,1999,第1-2卷]。所形成的部花青熱轉變為螺吡喃由於反-順再異構化階段而顯著延遲,這在著色形式恢復成初始的螺吡喃形式時發生且耗時數毫秒。
來自吲哚啉並螺吡喃產生著色形式相對較快,但由於熱化學及光化學降解而僅維持較少個數循環,而且對空氣中的氧也無抵抗力[v.malatesta,photodegradationoforganicphotochromesinorganicphotochromicandthermochromiccompounds.第2卷,j.c.crano和r.j.guglielmetti編。kluweracademic/plenumpublishers,newyork,1991,第473頁]。除此之外,部花青形式的螺吡喃可易於水解[stafforst,t.和hilvert,d.kineticcharacterizationofspiropyransinaqueousmedia.chem.commun.2009,287-288]。
新的一類光致變色化合物,即6-硝基-1,3,3『,4-四氫螺[苯並吡喃-2,2『-吲哚],具有吲哚啉螺吡喃的碳骨架(其中吡喃環中的雙鍵被氫化),在上文提到的lithuanian專利lt6024(wo2014/035244)中描述。
所述1『,3,3『,4-四氫螺[苯並吡喃-2,2『-吲哚](a)在受uv輻射輻射之後,形成著色的4-硝基酚鹽片段(b),其由於分子熱運動而在數十納秒內恢復至初始狀態。這是由於不存在雙鍵,因此不必要從反異構形式異構化為順異構形式。然而,化合物a對鹼性介質極為敏感。在不對稱的碳原子處進行直接取代且形成具有4-硝基苯酚片段的著色化合物c,如果溶液中存在任意量的氫氧根離子,光致變色作用則消失。
因此,重要的是具有反應較快且在鹼性溶液中不喪失其光致變色特性的光致變色化合物。
附圖簡要說明
本發明由以下圖示進行說明,其中:
圖1展示化合物6a在添加naoh(1mmolnaoh)後的乙腈中的動力學光譜;
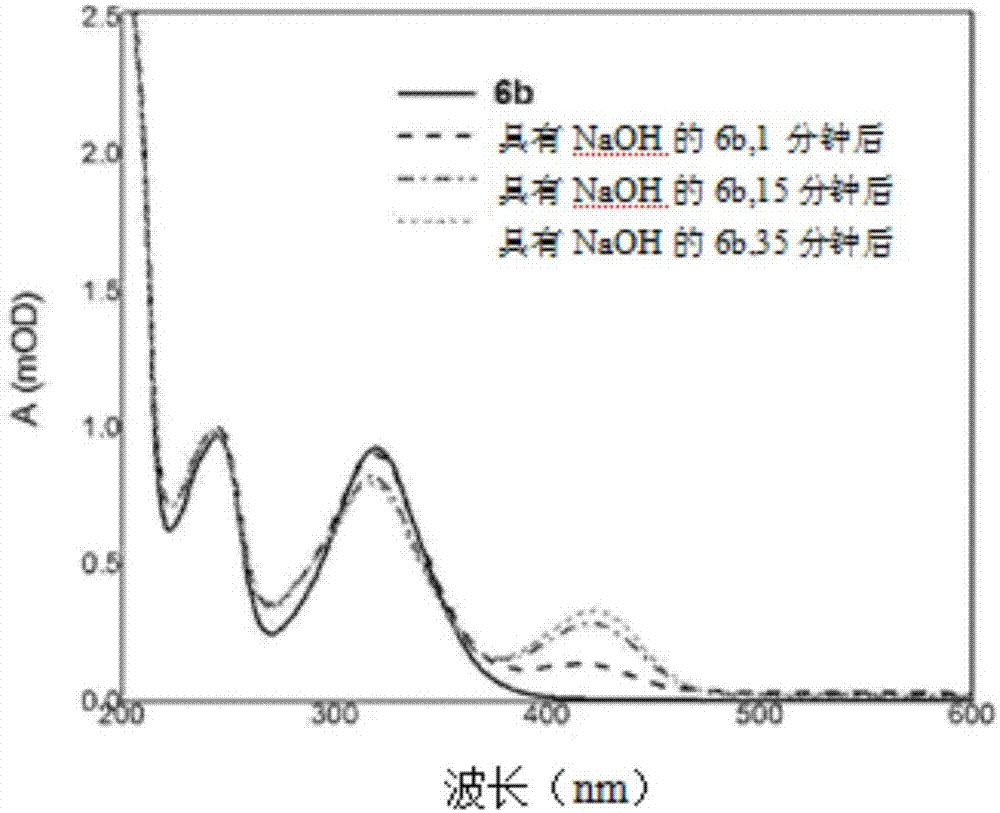
圖2展示化合物6b在添加naoh(1mmolnaoh)後的乙腈中的動力學光譜。
技術實現要素:
本發明的目標在於獲得新的光致變色化合物,它們通過甲醇苯並[f][1,3]氧氮雜卓並[3,2-a]吲哚中的3,4-二氫吡喃環的打開和閉合來起作用,其打開/閉合的速率高於已知光致變色體的速率,而且在中性和鹼性介質中都具有活性。
為實現此目標,本發明提出通式(i)和(ii)的化合物
其中
r1和r2各自獨立地為甲基、乙基或為螺環的部分:-(ch2)4-、-(ch2)5-;
r3為c1-c6-烷基、ω-官能化烷基、苄基、苯基、烯丙基。
此處,ω-官能化烷基具有可聚合基團(乙烯基、丙烯酸酯基、環氧基)或硫醇基,能連接至各種基質。
更具體而言,本發明涵蓋通式(i)或(ii)之化合物,其中r1和r2為甲基且r3為乙基或苄基。
本發明的優選目標化合物為:
(5ar*,12r*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓-12-甲醯胺(6a);
(5ar*,12s*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓-12-甲醯胺(7);
(5ar*,12r*,13s*)-n-苄基-6,6,8-三甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓-12-甲醯胺(6b)。
本發明的目標化合物的特徵在於光致變色特性。其可用作光致變色開關。
本發明的另一目標為通式(5)的化合物,
其中r3為乙基或苄基,且所述化合物為用於獲得本發明的光致變色化合物的中間物。
本發明的又一目標為通式(4)的化合物,其用於合成化合物(5):
其中r3為乙基或苄基。
本發明的另一目標為式(3)的化合物,其用於合成化合物(4):
本發明的又一目標為式(2)的3h-吲哚鹽,其用於合成式(3)的化合物:
具體實施方式
研發出新種類的光致變色化合物,包含通式(i)和(ii)的衍生物,順-和反-甲醇苯並[f][1,3]氧氮雜卓並[3,2-a]吲哚衍生物
與最近公開的光致變色化合物1',3',3'-三甲基-6-硝基-1',3,3',4-四氫螺[苯並吡喃-2,2'-吲哚]iii和iv類似,這些衍生物具有由吲哚(a)和色滿(b)雜環系統形成的1',3,3',4-四氫螺[苯並吡喃-2,2'-吲哚]的骨架。
然而,本發明的化合物帶有另一c-c鍵(上文中以黑體顯示),其降低了吡喃環的構象不穩定性。
發現本發明化合物的結構的獨特之處在於在紫外線輻射對i型化合物之作用下,3,4-二氫[2h]吡喃環中之c-o鍵發生斷裂,產生著色的兩性離子v,其具有著色的4-硝基酚鹽片段。此片段僅通過一個鍵連接,能繞其轉動。同時,當已知的高速光致變色體a被uv輻射觸發時,產生兩性離子化合物b,其中4-硝基酚鹽部分通過三個可能繞著旋轉的單鍵與吲哚鹽部分連接。與結構b(3個鍵)相比,在結構v(1個鍵)中由於可能繞c-c單鍵旋轉而導致自由度降低的有利之處在於二氫吡喃環比5ns更快地恢復,而且化合物v恢復為初始結構i。
如上文提到的,本發明包括對於獲得目標化合物而言必要的新穎中間物。
在發明人的知識範圍內,未描述中間物(2)、(3)、(4)、(5),但它們可以通過已知的合成方法來製備。通過使通式(4)的化合物例如與2-羥基-5-硝基苯甲醛縮合獲得通式(5)的中間物。通過用碘乙烷或苄基氯使7-甲氧基-9,9,9a-三甲基-9h-咪唑並[1,2-a]吲哚-2-酮烷基化來製備通式(4)的化合物。通過使3h-吲哚鹽環化來獲得式(3)的化合物,且通過用氯乙醯胺使2,3,3a-三甲基-5-甲氧基-3h-吲哚烷基化來獲得式(2)的後一種鹽。對於有機合成中的技術人員而言應了解如何來製備本發明的其它化合物。
通過處理發現,當本發明中提議的化合物的溶液被單色紫外線(例如,nd:yag雷射(eksplanl30);三次諧波;波長355nm;脈衝持續時間-5ns;脈衝能量3mj)輻射時,連接具有氧原子的手性中心(*標記)的鍵發生異裂,導致形成兩性離子化合物,包含連接3h-吲哚鹽陽離子和著色4-硝基酚鹽陰離子的片段的兩個碳原子的構象限制鏈。在切斷輻射之後,著色形式的化合物熱恢復至初始的無色狀態,之後酚鹽陰離子的氧原子連接至吲哚環系統的α-碳原子。本發明的化合物甲醇苯並[f][1,3]氧氮雜卓並[3,2-a]吲哚的特徵在於環閉合/打開的總速度較高,除此之外它們也在氧環境中進行操作。順-甲醇苯並[f][1,3]氧氮雜卓並[3,2-a]吲哚對環境的ph的敏感性較小,尤其對於鹼性介質而言,而6-硝基-1',3,3',4-四氫螺[苯並吡喃-2,2'-吲哚]對鹼極為敏感。本專利申請中描述的化合物通過在回流下用乙醇中的氫氧化鉀進行處理使相應的吲哚啉並螺吡喃發生再環化反應來合成。
所述甲醇苯並[f][1,3]氧氮雜卓並[3,2-a]吲哚可根據下文的通用流程來獲得:
實施本發明的方式
下文提供關於實際實施方案實例的信息,公開本發明化合物的製備方法及其特性。提供此信息僅出於說明目的而提供且不限制本發明的範疇。
實施例1
5-甲氧基-2,3,3-三甲基-3h-氯化吲哚鹽(初始化合物)
將5-甲氧基-2,3,3-三甲基-3h-吲哚12g(0,063mol)溶解於甲苯(20ml)中,將冷凝器連接至燒瓶且逐滴添加乙醯氯9ml(ρ=1.104g/ml,9,94g)。在將混合物冷卻至周圍溫度後,將混合物在5℃下儲存12h。過濾形成的晶體、用丙酮和冷的乙醚洗滌。產量為6.6g微黃色晶體狀的氯化吲哚鹽(46.12%)。熔點201-203℃。1hnmr(400mhz,dmso-d6):δ1.45(s,6h,2×ch3),2.62(s,3h,2-ch3),3.82(s,3h,5-och3),7.02(dd,j=8.8hz,j=2.4hz,1h,6-h),7.37(d,j=2.4hz,1h,4-h),7.54(d,j=8.4hz,1h,7-h)。13cbmr(100mhz,dmso-d6):δ14.3,22.0(2×c),53.8,55.9,109.4,113.9,117.2,134.9,145.3,159.7,193.0.ir(kbr),ν(cm-1):3134(n-h),3021,2973,1623,1479,1404,1289,1022,797。
實施例2
1-胺甲醯基甲基-5-甲氧基-2,3,3-三甲基[3h]氯化吲哚鹽(中間化合物2)
將根據實施例1獲得的5-甲氧基-2,3,3-三甲基-3h-氯化吲哚鹽(6g,0.026mol)溶解於最小量的水中、用碳酸鈉中和並用乙醚(2×30ml)萃取。將合併的萃取物經無水硫酸鈉乾燥且在旋轉蒸發儀上除去溶劑。向獲得的殘餘物(4.8g,0,025mol)中添加α-氯乙醯胺(2.6g,0.027mol)並將混合物在140℃下用冷凝器在鄰二甲苯(7ml)中加熱2h。冷卻後,過濾形成的晶體並用冷的乙醚洗滌。從乙醇再結晶後,獲得5g產物,產率為69.7%的深棕色晶體,熔點240-242℃。1hnmr(400mhz,dmso-d6):δ1.54(s,6h,2×ch3),2.73(s,3h,ch3),3.85(s,3h,o-ch3),5.36(s,2h,ch2co),7.14(dd,j=8.8hz,j=2.4hz,1h,6-h),7.51(d,j=2.4hz,1h,4-h),7.79-7.81(m,2h,7-h,1/2nh2),8.51(pl.s,1h,1/2nh2)。13cbmr(100mmhz,dmso-d6):δ14.4,22.6(2×c),50.1,54.5,56.6,109.9,114.9,116.3,135.2,143.9,161.1,165.1,196.4.ir(kbr),ν(cm-1):3290(n-h),3097,2977,1689(c=o),1298,1030,833。
實施例3
7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮(中間化合物3)
將根據實施例2的方法合成的1-胺甲醯基甲基-5-甲氧基-2,3,3-三甲基-3h-氯化吲哚鹽(5g,0,018mol)溶解於最小量的水中、用碳酸鈉中和並用乙醚(2×25ml)萃取。將醚萃取物合併、經無水硫酸鈉乾燥並在旋轉蒸發儀上蒸發溶劑。向獲得的中和混合物(5g,0,02mmol)中添加乙醇(20ml)和冰醋酸(9,2ml)並將混合物在100℃下加熱1h。在將混合物冷卻後,將其倒入水(50ml)中並用na2co3中和,直到開始形成白色晶體。將物質過濾、用水洗滌並從乙醇再結晶。產物的產量為2.5g(50%),呈白色晶體狀,熔點為209-211℃。1hnmr(400mhz,cdcl3):δ1.17(s,3h,ch3),1.36(s,3h,ch3),1.50(s,3h,ch3),3.76(s,3h,o-ch3),3.77(ab-q,j=16.4hz,2h,ch2),6.62(d,j=2.8hz,1h,4-h),6.64(d,j=8.8hz,1h,7-h),6.70(dd,j=8.8hz,j=2.4hz,1h,6-h),7.86(pl.s,1h,nh)。13cbmr(100mhz,cdcl3):δ21.2,22.3,27.5,47.0,55.8,55.9,92.0,109.1,112.8,113.3,140.2,144.3,155.9,174.9(c=o)。ir(kbr,cm-1):3333(n-h),3056,2975,1700(c=o)。msm/z(%):247(m+h+,100)。
實施例4
1-乙基-7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮(中間化合物4a)
將根據實施例3所述的方法合成的7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮2,5g(0,01mol)溶解於二甲基甲醯胺(25ml)中並向此溶液中添加新鮮粉末狀的koh(0,85g)。在鹼溶解之後,逐滴添加碘乙烷(2,45ml)並繼續攪拌3h。將混合物倒入水中並用乙醚(3×20ml)萃取。將醚萃取物合併、用5%的hcl溶液洗滌。用na2co3溶液中和、經無水硫酸鈉乾燥且在旋轉蒸發儀上蒸發溶劑。產物產量為2.3g(82.7%),呈微黃色樹脂狀物質。1hnmr(400mhz,cdcl3):δ1.04(s,3h,ch3),1.29(t,j=7.2hz,3h,ch2ch3),1.40(s,3h,ch3),1.47(s,3h,ch3),2.98-3.07(m,1h,1/2ch2ch3),3.63-3.72(m,1h,1/2ch2ch3),3.76(s,3h,och3),3.83(ab-q,j=15.2hz,2h,co-ch2),6.59(d,j=2.8hz,1h,4-h),6.70-6.71(m,2h,ar-h)。13cnmr(100mhz,cdcl3):δ14.33,23.07,24.11,27.02,29.19,37.23,50.06,55.84,92.69,108.99,113.01,115.36,142.23,142.99,156.05,171.22(c=o)。ir(kbr),ν(cm-1):3029,2984,1703(c=o),1491,1273,1014,816。
實施例5
1-苄基-7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮(中間化合物4b)
將根據實施例3所述的方法合成的7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮2,0g(0,008mol)溶解於二甲基甲醯胺(18ml)中且向此溶液中添加0,7g(0,12mol)新鮮研磨的koh。在鹼溶解之後,逐滴添加3.08苄基氯2.8ml(0,024mol,ρ=1.100g/ml,3.08g)且在室溫下繼續攪拌3hrs。將混合物倒入水中並用乙醚(3×20ml)萃取。將合併的萃取物用5%的hcl溶液洗滌並用na2co3溶液中和。將溶劑經無水硫酸鈉乾燥並用旋轉蒸發儀蒸發。通過柱色譜(洗提液:己烷/丙酮3:1)純化獲得的樹脂。產物的產量為1.4g(51.8%),呈淺橙色晶體狀,熔點122-123℃。1hnmr(400mhz,cdcl3):δ1.01(s,3h,9a-ch3),1.25(s,3h,9-ch3),1.37(s,3h,9-ch3),3.73(ab-d,j=15.2hz,1h,1/2ch2co),3.73(s,3h,7-och3),4.14(dd,j=15.5hz,j=5.8hz,2h,ch2-ph),4.95(ab-d,j=15.6hz,1h,1/2ch2co),6.55(d,j=2.5hz,1h,8-h),6.69(dd,j=8.4hz,j=2.5hz,1h,6-h),6.74(d,j=8.4hz,1h,5-h),7.21-7.28(m,5h,ar-h)。13cnmr(100mhz,cdcl3):δ23.3,23.9,28.7,45.4,50.1,55.7,55.8,93.1,108.9,113.0,115.6,127.5,127.6(2×c),128.7(2×c),137.7,142.1,143.2,156.2,172.1(c=o)。ir(kbr),ν(cm-1):3023,2969,1706(c=o),1493,1278,1027,730。
實施例6
n-乙基-2-(5'-甲氧基-3',3'-二甲基-6-硝基螺[苯並吡喃-2,2'-吲哚基]-1'(3'h)-基)乙醯胺(中間化合物5a)
將根據實施例4所述的方法合成的1-乙基-7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮2,5g(0.009mol)和2-羥基-5-硝基苯甲醛1.52g(0.009mol)添加至冰醋酸(20ml)中並在100℃下加熱3hrs。反應完成後,將混合物倒入5%的醋酸鈉溶液(250ml)中並用醋酸乙酯(2×30ml)萃取。將合併的萃取物經無水硫酸鈉乾燥並在旋轉蒸發儀上除去溶劑。通過柱色譜(洗提液:己烷/丙酮3:1)純化獲得的樹脂。產物的產量為1.3g(33.8%),呈紫色非晶物質狀。熔點為95-96℃。1hnmr(400mhz,cdcl3):δ1.09(t,j=7.2hz,3h,ch2ch3),1.26(s,3h,3』-ch3),1.3(s,3h,3』-ch3),3.25-3.34(m,2h,ch2ch3),3.59(ab-d,j=17.6hz,1h,1/2ch2co),3.80(s,3h,5』-och3),3.87(ab-d,j=17.6hz,1h,1/2ch2co),5.83(d,j=10.4hz,1h,ch=ch),6.43(d,j=8.4hz,1h,7』-h),6.49(t,j=5.2hz,1h,nh),6.72(dd,j=8.4hz,j=2.8hz,1h,6』-h),6.75-6.78(m,2h,4-h,4』-h),6.94(d,j=10.4hz,1h,ch=ch),8.00(d,j=2.8hz,1h,7-h),8.03(dd,j=8.8hz,j=2.8hz,1h,5-h)。13cnmr(100mhz,cdcl3):δ15.0,20.1,26.1,27.0,34.3,48.8,52.9,56.0,106.6(螺-c),108.3,110.0,111.7,115.6,118.3,120.4,123.1,126.3,129.4,137.8,140.0,141.5,155.4,158.8,169.3(c=o)。ir(kbr),ν(cm-1):3403(n-h),3065,2966,1666(c=o),1520,1481,1337,1273,1089,951。
實施例7
n-苄基-2-(5'-甲氧基-3',3'-二甲基-6-硝基螺[苯並吡喃-2,2'-吲哚基]-1'(3'h)-基)乙醯胺(中間化合物5b)
將根據實施例5所述的方法合成的1-苄基-7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮1.0g(0.003mol)和2-羥基-5-硝基苯甲醛(0.5g,0.003mol)添加至冰醋酸(10ml)中並在100℃下加熱3hrs。反應完成後,將混合物倒入5%的醋酸鈉溶液(100ml)中並用醋酸乙酯(2×30ml)萃取。將合併的萃取物經無水硫酸鈉乾燥並在旋轉蒸發儀上除去溶劑。通過柱色譜(洗提液:己烷/丙酮3:1)純化獲得的樹脂。產物的產量為0.41g(28.7%),呈紫色非晶物質狀。熔點為84-85℃。1hnmr(400mhz,cdcl3):δ1.18(s,3h,3』-ch3),1.27(s,3h,3』-ch3),3.71(ab-d,j=17.6hz,1h,1/2ch2co),3.80(s,3h,5』-och3),3.91,(ab-d,j=17.6hz,1h,1/2ch2co),4.45(d,j=5.6hz,2h,ch2-ph),5.76(d,j=10.2hz,1h,ch=ch),6.45(d,j=8.0hz,1h,4-h),6.60(d,j=8.4hz,1h,6』-h),6.71(dd,j=8.4hz,j=2.4hz,1h,7』-h),6.74(d,j=2.4hz,1h,4』-h),6.83(t,j=5.8hz,1h,nh),6.92(d,j=10.2hz,1h,ch=ch),7.14-7.17(m,2h,ar-h),7.27-7.29(m,3h,ar-h),7.96-7.80(m,2h,5-h,7-h)。13cnmr(100mhz,cdcl3):δ20.14,26.10,43.43,48.75,52.97,56.06,106.65(螺-c),108.23,110.06,111.79,115.61,118.29,120.51,123.03,126.31,127.57(2×c),127.72,128.87(2×c),129.41,137.66,138.0,139.84,141.58,155.38,158.66,169.49(c=o)。ir(kbr),ν(cm-1):3395(n-h),3030,2930,1671(c=o),1519,1494,1481,1337,1272,1089,951。
實施例8
(5ar*,12s*,13s*)-和(5ar*,12r*,13s*)-n-乙基-8-甲氧基-6,6-二甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓並-12-甲醯胺(目標化合物6a、7)
將根據實施例6所述的方法合成的螺化合物5a(1g,0.002mol)溶解於乙醇(30ml)中,添加精細研磨的氫氧化鉀(0.34g,0.006mol)且使混合物回流2hrs。反應完成後,在旋轉蒸發儀上除去大部分的溶劑,並向殘餘物中逐滴添加水直到溶液變得渾濁。使混合物在5℃下保持10hrs。過濾形成的晶體、用小量冷的乙醚洗滌並從乙醇再結晶。將濾液用醋酸乙酯(2×30ml)萃取,將合併的萃取物經無水硫酸鈉乾燥並在旋轉蒸發儀上除去溶劑。通過柱色譜(洗提液:己烷/丙酮5:1)純化殘餘物。使獲得的產物自乙醇再結晶。
順-異構體(6a)
產率10.4%,黃色晶體,熔點241-242℃。1hnmr(400mhz,cdcl3):δ0.61(t,j=7.2hz,3h,ch2ch3),1.48(s,3h,6-ch3),1.50(s,3h,6-ch3),2.16(d,j=11.6hz,1h,14-ha),2.20(dd,j=11.6hz,j=3.6hz,1h,14-hb),2.81-2.91(m,1h,1/2ch2ch3),3.12-3.23(m,1h,1/2ch2ch3),3.78(s,3h,8-och3),3.80(t,j=3.6hz,13-h),3.88(d,j=4.8hz,1h,12-h),6.43(d,j=9.2hz,1h,ar-h),6.66-6.69(m,2h,ar-h),6.89-6.92(m,2h,ar-h,nh),7.98(d,j=2.8hz,1h,1-h),8.07(dd,j=9.2hz,j=2.8hz,1h,3-h)。13cnmr(100mhz,cdcl3):δ14.7,23.11,26.6,32.9,33.7,42.4,45.5,56.0,78.2,109.6,111.1,111.5,113.1,116.3,124.7,125.1,125.9,139.8,141.4,142.4,156.0,158.4,169.5(c=o)。ir(kbr),ν(cm-1):3288(n-h),3071,2982,1652(c=o),1513,1344,1269,1090,816。
反-異構體(7)
產率2.5%,微黃色晶體,熔點184-185℃。1hnmr(400mhz,cdcl3):δ1.09(t,j=7.2hz,3h,ch2ch3),1.37(s,3h,6-ch3),1.60(s,3h,6-ch3),2.05(d,j=11.6hz,1h,14-ha),2.69(dd,j=11.6hz,j=4.0hz,1h,14-hb),3.24-3.35(m,2h,ch2ch3),3.75(s,3h,8-och3),3.77(d,j=4.0hz,1h,13-h),4.31(s,1h,12-h),5.97(t,j=5.2hz,1h,nh),6.25(d,j=8.4hz,1h,10-h),6.59(dd,j=8.4hz,j=2.4hz,1h,9-h),6.75(d,j=2.4hz,1h,7-h),6.79(d,j=9.2hz,1h,4-h),8.03(dd,j=8.8hz,j=2.8hz,1h,3-h),8.16(d,j=2.8hz,1h,1-h)。ir(kbr),ν(cm-1):3298(n-h),3082,2970,1650(c=o),1510,1496,1338,1268,1086,917。
實施例9
(5ar*,12s*,13s*)-n-苄基-8-甲氧基-6,6-二甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓並-12-甲醯胺(目標化合物6b)
將根據實施例7所述的方法合成的螺化合物5b0.385g(0.8mmol)溶解於乙醇(10ml)中,添加精細研磨的氫氧化鉀(0.13g,2.4mmol)且使混合物回流2hrs。反應完成後,在旋轉蒸發儀上除去大部分的溶劑,並逐滴添加水直到溶液變得渾濁。使混合物在5℃下保持10hrs。過濾形成的晶體、用小量冷的乙醚洗滌並從乙醇再結晶。產率為23.4%,呈黃色晶體狀。熔點218-219℃。1hnmr(400mhz,cdcl3):δ1.46(s,6h,2×6-ch3),2.13(d,j=11.6hz,1h,14-ha),2.21(dd,j=11.6hz,j=4.0hz,1h,14-hb),3.78(s,3h,8-och3),3.82(t,j=4.0hz,1h,13-h),3.93-3.97(m,2h,1/2ch2-ph,12-h),4.51(dd,j=14.8hz,j=8.0hz,1h,1/2ch2-ph),6.46(d,j=9.2hz,1h,4-h),6.68-6.70(m,2h,ar-h),6.73-6.75(m,2h,ar-h),6.79(d,j=8.8hz,1h,ar-h),7.10-7.16(m,3h,ar-h),7.28-7.31(m,1h,nh),7.93(d,j=2.8hz,1h,1-h),7.97(dd,j=9.2hz,j=2.8hz,1h,3-h)。13cnmr(100mhz,cdcl3):δ23.0,26.6,33.0,42.1,42.8,45.4,55.9,78.3,109.6,111.1,111.3,113.0,116.3,124.6,125.3,125.5,127.4(2×c),127.5,128.5(2×c),137.7,139.8,141.3,142.4,156.0,158.2,169.5(c=o)。ir(kbr),ν(cm-1):3377(n-h),3029,2975,1676(c=o),1517,1496,1340,1262,1086,909。
由乙腈中的溶液(10-5m/l)確定目標化合物6a、b和7的光致變色特性。在掃描分光計(shimadzuuv-3101pc)上記錄穩定態的uv-可見光吸收光譜。用nd:yag雷射3次諧波輻射(eksplanl30,脈衝能量為每次脈衝3mj,λ=355nm,脈衝持續時間5ns)進行閃光光解實驗。通過對脈衝計數來確定脈衝數,同時開放形式的光吸收光譜中的最大值達到初始吸收最大值的0.8。使用光學上根據激發波長進行調整的苯甲酮在mecn中的溶液(0.5od,在355nm下)來測量量子產率。根據能態特徵(不同吸光度信號的生長相對於激發脈衝能量)和摩爾消光係數,與對照溶液相比來評估光致變色轉變成開放形式的量子產率。
表中提供測量結果。
表中呈現的關於光致變色特性的數據表明,本發明的化合物可用作分子光開關,其中在uv輻射下,發生3,4-二氫-2h-吡喃環打開且產生變色形式的化合物,接著熱恢復成初始的未變色形式。形成變色形式並恢復回未變色形式的總的持續時間小於5ns。這個過程可以重複多次。本發明的化合物的特徵在於光穩定性高,在操作5000次以上循環時無明顯的降解。
本發明的化合物對鹼性介質比早前提到的化合物a較不敏感。如果我們向類似物a在乙腈中的溶液中滴加氫氧化鈉溶液,氫氧根離子將立即攻擊α-c碳原子,3,4-二氫吡喃環將打開並且形成著色的硝基酚鹽。
同時,圖1中提供的光譜表明,在用氫氧化鈉處理化合物6a之後,3,4-二氫吡喃環保持穩定而且不形成著色的4-硝基酚鹽陰離子。化合物6b(圖2)對鹼性介質更敏感,但不像化合物a那麼敏感。圖2中提供化合物6b的動力學uv-可見光光譜。
總的來說,本發明與已知的現有技術相比具有以下主要優勢:
1.本發明的順-和反-甲醇苯並[f][1,3]氧氮雜卓並[3,2-a]吲哚形成著色形式以及恢復回初始的未著色狀態的總的持續時間比吲哚啉並螺吡喃或1',3,3',4-四氫螺[苯並吡喃-2,2'-吲哚]中明顯較短。
2.順-甲醇苯並[f][1,3]氧氮雜卓並[3,2-a]吲哚的特徵在於對介質變化尤其具有抗性,而且只有通過uv輻射的作用才會出現著色形式。鹼類不易於打開[1,3]苯並惡嗪環而且不產生著色的4-硝基酚片段。
3.本發明的化合物的特徵在於比1',3,3',4-四氫螺[苯並吡喃-2,2'-吲哚]對光破壞的抗性更高,而且可以承受5000次以上的打開-閉合循環。
4.這些化合物對空氣中的氧不敏感,而且可以用在未除去氧的環境中。
權利要求書(按照條約第19條的修改)
1.一種通式(i)或(ii)的化合物
其中
r1和r2各自獨立地為c1-c3-烷基或r1與r2一起形成-(ch2)4-、-(ch2)5-;
r3為c1-c6烷基、苄基、苯基、烯丙基。
2.根據權利要求1的通式(i)或(ii)的化合物,其中
r1為甲基、r2為甲基且r3為乙基或苄基。
3.根據權利要求1或2的化合物,其為(5ar*,12r*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓-12-甲醯胺(6a)。
4.根據權利要求1或2的化合物,其為(5ar*,12s*,13s*)-n-乙基-6,6,8-三甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓-12-甲醯胺(7)。
5.根據權利要求1或2的化合物,其為(5ar*,12s*,13s*)-(n-苄基-6,6,8-三甲基-2-硝基-12,13-二氫-6h-5a,13-甲醇吲哚並[2,1-b][1,3]苯並氧氮雜卓-12-甲醯胺(6b)。
6.根據前述權利要求中任一項的化合物,其展示光致變色特性,其特徵弛豫時間小於5ns。
7.一種根據權利要求1-6中任一項的化合物的用途,其用作分子光開關。
8.一種通式(5)的化合物,
其中r3為乙基或苄基。
9.一種通式(4)的化合物,
其中r3為乙基或苄基。
10.一種式(3)的7-甲氧基-9,9,9a-三甲基-9,9a-二氫-1h-咪唑並[1,2-a]吲哚-2(3h)-酮
11.一種式(2)的1-胺甲醯基甲基-5-甲氧基-2,3,3-三甲基[3h]氯化吲哚鹽