一種GaN-on-Si晶圓的製備方法與流程
2023-05-07 12:36:12 1
本發明屬於半導體領域,涉及一種GaN-on-Si的晶圓製備方法,尤其涉及一種利用MOCVD反應腔製備GaN-on-Si晶圓的方法。
背景技術:
隨著能效標準不斷提高,基於矽(Si)材料的功率器件的改進空間越來越小;人們將目光投向新材料領域,以期實現根本改進,引發新一代功率器件技術的革命性突破。
眾多新材料中,基於氮化鎵(GaN)的複合材料最引人關注。GaN基功率器件具有擊穿電壓高、電流密度大、開關速度快以及工作溫度高等優點,性能遠優於Si基器件;採用GaN基功率開關器件的電子系統效率可改善3.5~7%,體積可減小35%,兼具高性能和高可靠性。GaN外延材料可以選擇多種襯底,其中Si襯底上外延生長GaN具有明顯的成本優勢,一方面Si襯底的價格很低,另一方面Si襯底的尺寸較大(8英寸),能夠形成大規模生產的優勢。
目前製備矽基氮化鎵(GaN-on-Si)晶圓的主要方案是利用現有的MOCVD(金屬有機化學氣相沉積)設備來製備。
MOCVD設備作為一種典型的CVD設備,能夠提供在晶片(例如藍寶石外延片)表面生長用於發光的晶體結構GaN(氮化鎵)時所需的溫度、壓力和化學氣體組分等條件。MOCVD設備中設有真空的反應腔,反應腔中設有託盤,通過進氣裝置(例如噴淋頭)將反應氣體引入反應腔內,並輸送到放置在託盤上的晶片的表面進行處理,從而生長出特定的晶體結構例如GaN結構。
但是,現有的MOCVD設備主要是針對藍綠光的光電二極體(LED),其 特點是在一個反應腔內完成全部生長步驟,直至得到所需的材料結構為止。因此利用目前的MOCVD設備製備GaN-on-Si晶圓的方法均或多或少存在一些問題,如氮化鋁的生長速率較低、鋁利用效率較低、GaN-on-Si晶圓質量較差等。
技術實現要素:
針對上述現有技術中存在的問題,本發明提供了一種利用多個金屬有機化學氣相沉積裝置(MOCVD)反應腔分步驟製得GaN-on-Si晶圓的方法。
為達此目的,本發明採用以下技術方案:
第一方面,本發明提供了一種利用金屬有機化學氣相沉積裝置製備GaN-on-Si晶圓的方法,所述金屬有機化學氣相沉積裝置包括兩個不同的反應腔,具體包括以下步驟:
在第一反應腔內通過外延反應在矽基襯底上沉積AlN(氮化鋁)薄膜,再在AlN薄膜上沉積組成為AlxGa1-xN的第一緩衝層,其中A<X≤0.9,A為0.4~0.6;
將沉積有AlN薄膜和第一緩衝層的矽基襯底轉移至第二反應腔內;
在第二反應腔內通過外延反應進一步沉積組成為AlYGa1-YN的第二緩衝層,其中0.1≤Y≤A,A為0.4~0.6,然後在該第二緩衝層上沉積GaN層,製備得到GaN-on-Si晶圓。
其中,在沉積的組成為AlxGa1-xN的第一緩衝層中A<X≤0.9,X可為0.5、0.6、0.7、0.8或0.9等,A為0.4~0.6,例如0.4、0.5或0.6。
在沉積的組成為AlYGa1-YN的第二緩衝層中0.1≤Y≤A,Y可為0.1、0.2、0.3、0.4、0.5或0.6等,A為0.4~0.6,例如0.4、0.5或0.6。
作為本發明的優選方案,第一反應腔和第二反應腔均是金屬有機化學氣相 沉積反應腔,第一反應腔的高度小於第二反應腔;其中,所述的「高度」是指反應腔中噴淋頭下表面至託盤上表面之間的距離。
作為本發明的優選方案,第一反應腔高度為15~25mm,例如15mm、17mm、20mm、23mm或25mm等;第二反應腔高度為50~100mm,例如50mm、60mm、70mm、80mm、90mm或100mm等。
本發明根據製備得到的GaN-on-Si晶圓中AlGaN緩衝層中鋁組分含量的多少,將AlGaN緩衝層分為高鋁組分AlGaN緩衝層和低鋁組分AlGaN緩衝層,其中高鋁組分AlGaN緩衝層為組成為AlxGa1-xN的第一緩衝層,A<X≤0.9;低鋁組分AlGaN緩衝層為組成為AlYGa1-YN的第二緩衝層,0.1≤Y≤A,其中A為0.4~0.6。
第一反應腔的高度小於第二反應腔是由於在材料製備過程中,高鋁組分AlGaN緩衝層中的鋁有機源極易與氨氣發生預反應生成鋁團聚物,降低鋁的利用率,限制了AlN生長速率和AlGaN中的鋁摻雜效率,但是通過降低反應腔的高度,即縮短鋁有機源極易與氨氣混合距離,可以有效的抑制預反應,實現高的AlN生長速率和AlGaN中高的鋁摻雜。在材料製備過程中,低鋁組分AlGaN緩衝層中鋁有機源與氨氣的預反應隨著鋁含量減少而減弱,而材料反應中生成的易沉積於反應腔內壁的副產物對GaN-on-Si晶圓影響更為顯著,因而需要選擇合適的反應腔高度優化流場分布,抑制副產物的沉積,因而第二反應腔的高度要大於第一反應腔。
作為本發明的優選方案,第一反應腔和第二反應腔均是單片反應腔。
作為本發明的優選方案,第一反應腔和第二反應腔均支持託盤轉速為500轉以上/分鐘的高速旋轉。
作為本發明的優選方案,將矽基襯底轉移至第二反應腔的過程是在密閉真空環境下完成的。
作為本發明的優選方案,在第一反應腔內沉積有AlN薄膜和第一緩衝層的的矽基襯底溫度降至400~500℃後,將該矽基襯底轉移至第二反應腔內,其中降溫溫度可為400℃、430℃、450℃、470℃或500℃等。
作為本發明的優選方案,在第一反應腔內沉積AlN薄膜和沉積第一緩衝層的溫度為1150~1250℃,例如1150℃、1170℃、1200℃、1230℃或1250℃等;壓力為45~55mbar,例如45mbar、47mbar、50mbar、53mbar或55mbar等。
優選地,在第二反應腔內中沉積第二緩衝層和GaN層的溫度為1050~1150℃,例如1050℃、1070℃、1100℃、1130℃或1150℃等;壓力為100~200mbar,例如100mbar、130mbar、150mbar、170mbar或200mbar等。
作為本發明的優選方案,在第二反應腔沉積第二緩衝層和GaN層的同時,在第一反應腔內在另一批次矽基襯底上沉積AlN薄膜和第一緩衝層。
作為本發明的優選方案,所述方法包括以下步驟:
在第一反應腔內通過外延反應在矽基襯底上沉積AlN薄膜,再在AlN薄膜上沉積組成為AlxGa1-xN的第一緩衝層,其中A<X≤0.9,A為0.4~0.6,沉積溫度為1150~1250℃,壓力為45~55mbar;
在第一反應腔內沉積有AlN薄膜和第一緩衝層的矽基襯底溫度降至400~500℃後,將該矽基襯底在密閉真空環境下轉移至第二反應腔內;
在第二反應腔內通過外延反應進一步沉積組成為AlYGa1-YN的第二緩衝層,其中0.1≤Y≤A,A為0.4~0.6,在該緩衝層上沉積GaN層,沉積溫度為1050~1150℃,壓力為100~200mbar,製備得到GaN-on-Si晶圓。
與現有技術相比,本發明具有以下有益效果:
本發明所述的金屬有機化學氣相沉積裝置採用兩個反應腔外延生長GaN-on-Si晶圓,能有效地消除鎵回熔產生的晶體缺陷導致電性良品率低等問題,提高氮化鋁的生長速率和鋁利用效率;進一步地,還提高了工藝重複一致性和可靠性;此外,還能提高外延生長材料的均勻性和設備效率,有效提升產能和降低外延生產的成本。
附圖說明:
圖1是本發明所述製備GaN-on-Si晶圓的方法的工藝流程圖;
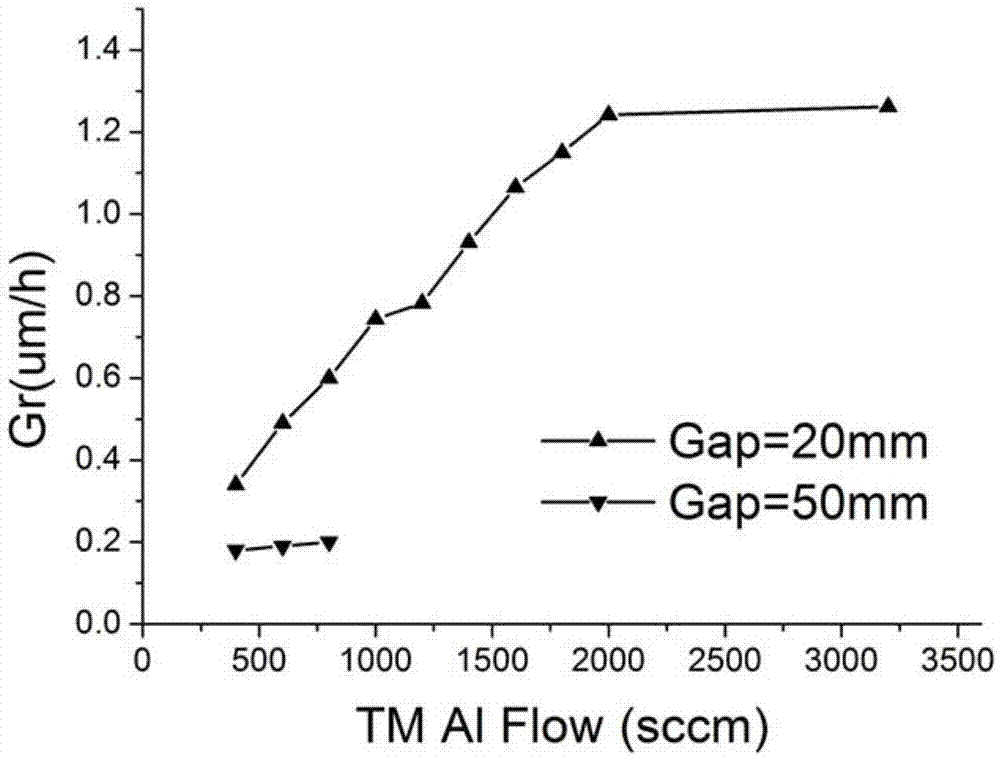
圖2是本發明實施例1中兩種MOCVD沉積AlN薄膜的生長速率隨TMAl流量的變化圖;
圖3是本發明實施例2中兩種MOCVD製備的AlGaN薄膜中實測鋁組分與理論鋁組分的對比關係圖,其中圖3a為實施例2中採用本發明所述方法製備的AlGaN薄膜中實測鋁組分與理論鋁組的對比關係圖,圖3b為採用傳統方法製備的AlGaN薄膜中實測鋁組分與理論鋁組的對比關係圖。
具體實施方式
以下結合若干個具體實施例,示例性說明及幫助進一步理解本發明,但實施例具體細節僅是為了說明本發明,並不代表本發明構思下全部技術方案,因此不應理解為對本發明總的技術方案限定,一些在技術人員看來,不偏離發明構思的非實質性改動,例如以具有相同或相似技術效果的技術特徵簡單改變或替換,均屬本發明保護範圍。
本申請發明人通過分析,在傳統的生產GaN-on-Si晶圓的方法中,由於在同一個反應腔內完成所有的外延步驟,會產生鎵回熔的現象,產生的晶體缺陷 導致電性良品率低。
針對傳統的生產GaN-on-Si晶圓的方法的不足,本申請具體實施例部分提供了一種利用金屬有機化學氣相沉積裝置製備GaN-on-Si晶圓的方法,該金屬有機化學氣相沉積裝置包括多個不同的反應腔,例如兩個,其具體流程如圖1所示:
首先,在第一反應腔內通過外延反應在矽基襯底上沉積AlN薄膜,再在AlN薄膜上沉積組成為AlxGa1-xN的第一緩衝層,其中A<X≤0.9,A為0.4~0.6,該過程沉積溫度為1150~1250℃,壓力為45~55mbar。
然後,在第一反應腔內沉積有AlN薄膜和第一緩衝層的矽基襯底溫度降至400~500℃後,將矽基襯底在密閉真空環境下轉移至第二反應腔內。
最後,在第二反應腔內通過外延反應進一步沉積組成為AlYGa1-YN的第二緩衝層,其中0.1≤Y≤A,A為0.4~0.6,在該第二緩衝層上沉積GaN層,製備得到GaN-on-Si晶圓,該過程沉積溫度為1050~1150℃,壓力為100~200mbar,製備得到GaN-on-Si晶圓。
進一步地,第一反應腔和第二反應腔均為MOCVD反應腔,且第一反應腔的高度小於第二反應腔,其中,「高度」是指反應腔中噴淋頭下表面至託盤上表面之間的距離。在一個可選的實施方案中,第一反應腔和第二反應腔均為單片反應腔。在另一個可選的實施方案中,第一反應腔和第二反應腔均支持高速旋轉,即反應腔中託盤的轉速為500rpm以上,即託盤轉速大於等於500轉/分鐘。進一步地,採用本實施例提供的方法,可以形成GaN-on-Si晶圓的流水製備。即在第二反應腔內在某一批次(一片或多片)矽基襯底上沉積第二緩衝層和GaN層的同時,在第一反應腔內在另一批次(一片或多片)矽基襯底上 沉積AlN薄膜和第一緩衝層,因而能有效提高GaN-on-Si晶圓生產能力。
實施例1:
本實施例提供了一種利用分布式MOCVD設備(即該MOCVD設備包括多個反應腔)製備GaN-on-Si晶圓中AlN方法,該方法包括如下步驟:
將裝有Si襯底置於分布式MOCVD設備的第一反應腔,該反應腔的高度為15~25mm,例如20mm,將溫度升至1200℃,壓力設定為50mbar,在Si襯底上沉積AlN薄膜,通過調節鋁有機源(TMAl)的流量,實時原位測量AlN薄膜的生長速率。
為了便於說明本發明實施例的優點,採取同樣的工藝又在傳統一步式MOCVD(在一個反應腔內完成全部外延步驟)中重複該實驗,由於傳統一步式MOCVD考慮到更多的外延步驟,反應腔的高度受到的限制因素更多,且對一個反應腔來說,反應腔高度一旦確定後通常不再能夠調整。現有的MOCVD設備中,特別是支持高速旋轉(500rpm以上,即500轉以上/分鐘)的MOCVD設備中,反應腔的高度一般設為50~100mm。
其中Si襯底層的尺寸為8英寸(200mm),厚度為1000μm。
兩種方法沉積AlN薄膜的生長速率隨TMAl流量的變化如圖2所示,圖2中Gap代表反應腔高度。
從圖2中可以看出,現有技術中,當反應腔的高度為50mm時,AlN的生長速率隨TMAl的流量緩慢增加,當TMAl流量大於800sccm(sccm為氣體流量單位,代表標準每立方釐米),生長速率飽和,AlN的最大生長速率僅為0.2μm/h。而本實施例中,當反應腔的高度為20mm時,AlN生長速率隨TMAl的流量線性增大,直到TMAl的流量大於2000sccm為止,AlN最大生長速率 可達1.26μm/h,顯著超出現有技術。
本實施例說明採用分布式MOCVD製備GaN-on-Si晶圓中,在第一反應腔即高度較小的反應腔沉積AlN的過程中,較現有技術中AlN的生長速率提高了,鋁的利用率提高了。
實施例2:
本實施例提供了一種利用分布式MOCVD製備GaN-on-Si晶圓中AlGaN方法,所述方法如下:
將裝有Si襯底置於分布式MOCVD設備的第一反應腔,該反應腔的高度為20mm,將溫度升至1200℃,壓力設定為50mbar,在Si襯底上沉積200nmAlN,然後繼續沉積400nm的AlGaN薄膜(第一緩衝層),通過調節鋁有機源和鎵有機源的比例,製備了不同鋁組分的3個樣品,然後取出測量樣品中的鋁組分。為了便於說明本實施例的優點,又在傳統一步式MOCVD(高度為50mm)中採取同樣的方法製備了另外三塊AlGaN樣品,待生長結束後取出測量樣品中的鋁組分。
兩種MOCVD製備的AlGaN薄膜中實測鋁組分與理論鋁組分的對比關係如圖3所示。從圖3a可以看出,採用分布式MOCVD的第一個反應腔在製備AlGaN特別是高鋁組分的AlGaN時,實測鋁組分約為理論鋁組分的80%。如圖3b所示,而傳統的一步式反應腔製備AlGaN,實測鋁組分約為理論鋁組分的50%。
本實施例說明採用分布式MOCVD製備GaN-on-Si晶圓中,在第一反應腔即高度較小的反應腔沉積高鋁組分AlGaN的過程中,較現有技術中AlGaN的鋁實際含量顯著提高,鋁的利用率提高了。
實施例3:
本實施例提供了一種利用MOCVD製備GaN-on-Si晶圓的方法,所述方法如下:
將裝有Si襯底置於MOCVD設備的第一反應腔,將溫度升至1150~1250℃,壓力設定為45~55mbar,例如將溫度升至1200℃,壓力設定為50mbar,在Si襯底上依次沉積AlN薄膜和第一緩衝層AlxGa1-xN(其中,A=0.5,0.5<X≤0.9),沉積完成後,待第一反應器溫度降至450℃後,將其轉移至第二反應腔,通常該轉移過程是在密閉真空環境下完成的。在第二反應腔中將溫度上升至1050~1150℃,壓力為100~200mbar,例如溫度設定為1100℃,設定壓力為150mbar,然後依次沉積第二緩衝層AlYGa1-YN(其中A=0.5,0.1≤Y≤0.5)和GaN層,製備得到GaN-on-Si晶圓。
製備得到的GaN-on-Si晶圓具有Si襯底層、AlGaN緩衝層和GaN層,其中Si襯底層的尺寸為8英寸,厚度為1000μm,Si襯底上有一層厚度為200nm的AlN薄膜,AlGaN緩衝層有兩層,第一層形成於AlN薄膜上,鋁組分含量為60wt%,厚度為1000nm,第二層形成於第一層AlGaN層上,鋁組分含量為40wt%,厚度為1000nm,GaN形成於第二AlGaN層上,厚度為1.5μm。
為了展示分反應腔分步驟製備GaN-on-Si晶圓方法的特點,又用傳統的方法,即採用一個反應腔製備了同樣結構的GaN-on-Si晶圓。
兩種方法製備的GaN-on-Si晶圓具體過程,採用反應腔分步驟方法製備GaN-on-Si晶圓的時間約為5小時,而傳統方法約為8小時,通過對比發現本發明實施例的方法可以顯著縮短晶圓的製備時間。
對比兩種方法製備GaN-on-Si晶圓的晶體質量,發現本發明實施例的方法 製備的GaN晶體質量也優於傳統的方法。衡量GaN晶體質量的參數為GaN(002)晶面和(102)面的X射線搖擺曲線的寬度,數值越小表明晶體質量越好。根據測試結果,本發明實施例的方法製備的GaN-on-Si晶圓中GaN(002)晶面和(102)面的X射線搖擺曲線的寬度分別為1000弧秒和2000弧秒,而採用傳統方法製備的GaN-on-Si晶圓中GaN(002)晶面和(102)面的X射線搖擺曲線的寬度分別為2000弧秒和3600弧秒。
綜合實施例1-3和對比例中的結果可以看出,本發明所述的金屬有機化學氣相沉積裝置採用兩個具有高度差的反應腔分步驟製備GaN-on-Si晶圓的方法:能有效地消除鎵回熔產生的晶體缺陷導致電性良品率低等問題;提高氮化鋁的生長速率、AlGaN中的鋁組分和鋁利用效率;縮短GaN-on-Si晶圓的製備時間;提高了GaN-on-Si晶圓的晶體質量。同時,該方法還可以提高了工藝重複一致性和可靠性,設備效率,有效提升產能和降低外延生產的成本,更符合大規模生產需要。
實施例4:
實施3中製備得到的GaN-on-Si晶圓若作為功率器件應用,還需要在GaN-on-Si晶圓上繼續沉積20nm的AlGaN,其中鋁組分的含量為20~30wt%。
實施例5:
實施例3中製備得到的GaN-on-Si晶圓若作為LED應用,還需要繼續在GaN-on-Si晶圓上繼續沉積n型GaN、InGaN/GaN超晶格和p型GaN。
申請人聲明,本發明通過上述實施例來說明本發明的詳細方法,但本發明並不局限於上述詳細方法,即不意味著本發明必須依賴上述詳細方法才能實施。所屬技術領域的技術人員應該明了,對本發明的任何改進,對本發明產品 各原料的等效替換及輔助成分的添加、具體方式的選擇等,均落在本發明的保護範圍和公開範圍之內。