一種無外源標記基因的抗PCV2轉基因豬製備方法與流程
2023-04-30 04:54:41 3
本發明涉及基因工程領域,特別涉及一種無外源標記基因的抗pcv2轉基因豬製備方法。
背景技術:
在家畜動物的生長及疾病防治中,轉基因技術日趨成熟,為便於轉基因細胞系的篩選及轉基因動物的檢測,在轉入目的基因的同時,常常同時轉入外源標記基因,而egfp綠色螢光蛋白可以在螢光顯微鏡下直接觀察,而常常作為一種標記基因;neo基因是一種抗生素抗性基因,細胞表達該基因可以滅活氨基糖甙類抗生素,破壞g418,而常常作為一種篩選標記基因用於轉基因單克隆細胞的篩選。因而這兩種基因無可避免的會出現在轉基因動物中,而轉基因動物成功獲得後這些標記基因便不再需要了。
同時,外源標記基因的存在將會對轉基因家畜的健康將產生不良的影響,且我國轉基因安全評價政策明文規定將標記基因等同於目的基因,同樣必須經過複雜的安全評價,這無疑直接影響了轉基因家畜的推廣和產業化進程。因此,在進行轉基因的操作中,通常在標記基因的兩側各加上一個相同的loxp序列,以便在富集轉基因成功的細胞後利用cre/loxp系統將轉基因細胞內多餘的標記基因刪除。
豬圓環病毒,屬於圓環病毒科,為單股環狀dna病毒,分為1型和2型。其中豬圓環病毒1型(pcv1)不致病,豬圓環病毒2型(pcv2)具有致病性。在養豬業疾病防控中,疫苗是人們用來對付疾病的最常用手段。而由於圓環病毒滅活疫苗與亞單位疫苗成本高、注射次數多和容易造成機體應激等缺點,以及免疫效力有待提高,並不能大規模的免疫接種。另外,疫苗的使用在控制由pcv2感染而引起pmws中獲得了一些成功,但是這種症候群的發病病源是多樣的,且在其他症候群上並沒有突破,所以通過轉基因手段引入抗pcv2sirna基因序列,使用rna幹擾技術對豬圓環病毒2型的防禦顯得更有實際價值。
在2013年12月25日公布的專利號為201310321355.6號,專利名稱為「抗豬圓環病毒2型的表達載體和轉基因豬及其構建方法」中公開了一種抗豬圓環病毒2型(pcv2)的表達載體和轉基因豬及其構建方法,並通過該方法獲得了f0代(原代)抗豬圓環病毒2型(pcv2)轉基因豬,該轉基因豬整合的外源基因包括目的基因抗pcv2sirna的基因序列及標記基因neo/egfp融合表達基因序列,而標記基因的存在,將對該轉基因豬產生不良的影響,並影響該轉基因豬的安全評價,不利於該轉基因豬的推廣及產業化進程。
技術實現要素:
本發明的目的是提供一種無外源標記基因的抗pcv2的轉基因豬製備方法,以解決上述問題。
根據本發明的一個方面,提供了一種無外源標記基因的抗pcv2轉基因豬製備方法,其是通過如下步驟來實現的:
(1)、抗pcv2表達載體的構建,該載體包含目的基因抗pcv2的sirna表達基因序列和標記基因序列;
(2)、篩選可穩定表達載體的單克隆轉基因細胞系;
(3)、將獲得的單克隆轉基因細胞系作為核供體細胞,進行體細胞核移植獲得f0代抗pcv2的轉基因豬;
(4)、將獲得的f0代抗pcv2的轉基因豬與母豬雜交繁育獲得能穩定遺傳的抗pcv2的f1代轉基因豬;
(5)、篩選f1代轉基因豬個體並進行外源基因整合位點分析;
(6)、將篩選出的f1代轉基因豬個體採取耳皮成纖維細胞,並進行耳皮成纖維細胞系的建立;
(7)、建立刪除了外源標記基因的細胞單克隆,並進行體細胞克隆獲得f2代無外源標記基因的抗pcv2的轉基因豬。
由此,通過上述方法獲得的抗pcv2的轉基因豬,可以特異表達抗pcv2sirna,對豬圓環病毒疾病起到防疫作用;同時該轉基因豬不含外源標記基因,不會對豬健康產生額外的負擔及影響,同時也可以簡化轉基因豬安全評價程序,有利於該轉基因豬的產業化推廣及應用。
在一些實施方式中,上述外源標記基因是neo/egfp融合表達基因,neo基因是一種抗生素抗性基因,細胞表達該基因可以滅活氨基糖甙類抗生素,破壞g418,而常常作為一種篩選標記基因用於轉基因單克隆細胞的篩選;而egfp為綠色螢光蛋白表達基因與目的基因共同轉染構建轉基因單克隆細胞系時,便於用顯微鏡直觀的觀察篩選結果,對轉基因動物也可以通過觀察綠色螢光蛋白的表達來初步判斷轉基因的結果,但egfp綠色螢光蛋白在細胞水平表達會對細胞產生一定的毒性,在轉基因豬個體中也會對豬的健康產生一定的影響,因此刪除了neo/egfp融合表達外源標記基因的轉基因豬會減少對該轉基因健康的影響,同時可以簡化轉基因豬安全評價程序,有利於該轉基因豬的產業化推廣及應用。
在一些實施方式中,f1代抗pcv2轉基因豬是通過f0代抗pcv2轉基因豬與同品種,且遺傳背景一致的野生型母豬雜交繁育而來。
在一些實施方式中,f0代抗pcv2轉基因豬與同品種,且遺傳背景一致的野生型母豬雜交繁育而來的f1代抗pcv2轉基因豬篩選的標準為高效特異表達抗pcv2sirna,且抗pcv2sirna基因序列的整合位點只有一個,由此篩選出來的f1代抗pcv2轉基因豬既可以穩定遺傳目的基因抗pcv2sirna,同時由於整合位點只有一個,便於定向刪除與目的基因同時整合在轉基因豬染色體上的外源標記基因。
在一些實施方式中,通過htncre重組酶來刪除外源表達基因的步驟如下:
(1)、復甦f1代抗pcv2轉基因豬耳皮成纖維細胞;
(2)、待細胞生長狀態良好時,用htncre重組酶處理細胞系;
(3)、24h後,對處理後的細胞系進行傳代培養,並在顯微鏡下挑選無綠色螢光表達的細胞單克隆團,並進行純化培養,即獲得無neo/egfp標記基因的細胞單克隆。
由此,通過htncre重組酶利用外源基因兩側的loxp位點,即可以刪除外源標記基因,且刪除標記基因後的細胞不表達綠色螢光,在細胞團中挑出不表達綠色螢光的細胞,進行純化建系,即可以獲得無neo/egfp標記基因的細胞單克隆。
在一些實施方式中,htncre重組酶處理細胞系刪除外源標記基因時,具體要求如下:待f1代抗pcv2轉基因豬耳皮成纖維細胞密度達80%左右時,用dpbs洗2次,加入opti-mem稀釋的htncre重組酶,使其終濃度為200ng/μl,孵育3h後吸走htncre工作液,用dpbs洗2次,加入10%fbs的完全培養基繼續培養細胞,由此可以獲得最佳的刪除效率,並獲得能保證刪除標記基因之後的細胞生長狀態較好。
在一些實施方式中,挑選無綠色螢光表達的細胞單克隆團的具體步驟如下:當經htncre重組酶處理的細胞培養至10天左右,細胞克隆團細胞數達到200個左右時,在倒置顯微鏡的觀察下於細胞培養皿底部圈出細胞克隆團所在位置,並在克隆團外圍再圈出無雜細胞的安全區域,同時用螢光顯微鏡觀察細胞克隆團有無螢光,做好標記,以確保挑取更純的無螢光細胞單克隆。由此,可以精確的挑選出無綠色螢光蛋白表達的細胞團,建系培養後,保證細胞系中無未刪除標記基因的雜細胞。
在一些實施方式中,獲得無neo/egfp標記基因的細胞單克隆的具體步驟如下:標記好無螢光細胞單克隆後,吸走細胞完全培養基,用dpbs洗2次,用鑷子將克隆環小心準確放在已做標記的細胞克隆上,每個克隆環內加入兩滴0.25%胰酶,在顯微鏡下觀察至細胞變圓時,加入50μl左右的fbs終止消化,用移液器小心吹起細胞並轉移至24孔板中,按24孔-6孔-60mm的順序進行傳代培養,以獲得無標記基因螢光表達的細胞單克隆。由此,可以保證細胞系無未刪除標記基因的雜細胞,且保證篩選出的細胞系生長狀態較好。
在一些實施方式中,刪除外源標記基因時htncre重組酶的終濃度還可以為100ng/μl,150ng/μl或300ng/μl等濃度,只是優選的選用200ng/μl。
上述的無外源標記基因的抗pcv2轉基因豬製備方法,具有如下有益效果:無外源標記基因的抗pcv2轉基因豬是通過構建含有目的基因(抗pcv2sirna基因)及外源標記基因(neo/egfp融合表達基因)的共表達載體,進行轉染製備轉基因單克隆細胞系,然後以此單克隆細胞系作為體細胞克隆技術的供核體細胞,來獲得含有目的基因及外源標記基因的f0代抗pcv2轉基因豬,然後通過f0代與品種、遺傳背景一致的野生型杜洛克母豬雜交繁育獲得能穩定遺傳的抗pcv2的f1代轉基因豬,篩選出抗pcv2sirna表達量高,且只有一個整合位點的f1代轉基因豬個體進行後續研究,採取該篩選出的f1代轉基因豬個體的耳皮成纖維細胞進行培養建系,對獲得的耳皮成纖維細胞系進行htncre重組酶處理,並篩選出刪除了標記基因的細胞單克隆,以此細胞單克隆作為體細胞克隆技術的供核細胞,進行體細胞克隆獲得f2代不含外源標記基因的抗pcv2轉基因豬,並對該f2代轉基因豬進行檢測,證實標記基因已被刪除,但抗pcv2sirna基因序列被保留。通過上述方法獲得的無外源標記基因的抗pcv2轉基因豬可以特異表達抗pcv2sirna,來抑制pcv2的表達及生物活性,從而起到防治豬感染圓環病毒疾病的作用。同時,該轉基因豬刪除了外源標記基因,減少了外源基因對轉基因豬個體健康的影響,同時也可以簡化轉基因豬安全評價程序,有利於該轉基因豬的產業化推廣及應用。
附圖說明
圖1為實施例1中抗pcv2轉基因豬f1代egfp外源標記基因綠色螢光表達情況;
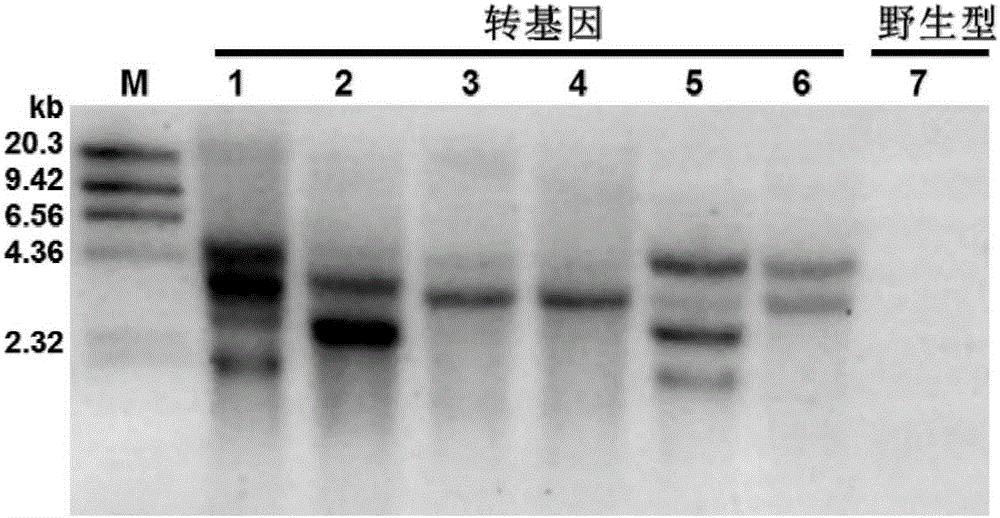
圖2為實施例2中抗pcv2(轉基因豬f1代的southernblot檢測結果,其中mw為dna分子量標準,transgenic為f1代抗pcv2轉基因豬樣品,wild-type為野生型對照豬樣品;
圖3為實施例3中不同f1代抗pcv2轉基因豬之間sirna相對表達情況,其中「3」標識tg3個體;
圖4為實施例4中f1代抗pcv2轉基因豬tg3個體目的基因整合位點測序對比圖;
圖5為實施例5中f1代抗pcv2轉基因豬tg3耳皮成纖維細胞系的建立結果圖;
圖6為實施例6中htncre重組酶的表達純化sds-page分析結果圖,其中mw為蛋白分子量標準;1~3、5為誘導的溶菌產物粗分;4為未誘導的溶菌產物粗分;6為誘導的溶菌產物上清;7為誘導的溶菌產物沉澱;8為純化後的產物;
圖7為實施例7中htncre處理f1代抗pcv2轉基因豬tg3細胞系前後流式細胞檢測螢光結果圖;
圖8為實施例7中f1代抗pcv2轉基因豬tg3細胞克隆刪除標記前後螢光表達檢測結果圖;
圖9為實施例8中刪除標記的轉基因豬tg3細胞單克隆的pcr檢測結果圖,其中mw為dnamarker;nagtive為陰性對照;w為水;wt為野生型豬;positive為陽性對照;p為pegfp-sh2質粒;-cre為未經htncre處理;mix為htncre處理後的混合細胞;c1~c7為7個細胞單克隆;
圖10為實施例8中htncre重組酶介導的刪除反應原理以及pcr產物(530bp)測序驗證結果圖;
圖11為實施例10中無外源標記基因的抗pcv2轉基因豬的egfp表達情況結果圖;
圖12為實施例11中無外源標記基因的抗pcv2轉基因豬的pcr檢測結果圖。
具體實施方式
下面結合附圖對發明作進一步詳細的說明。
如無特殊說明,以下實施例中所使用的試劑均來源於市售,操作方法為現有常規操作方法。
本發明中所指的f1代抗pcv2轉基因豬通過專利號為201310321355.6,專利名稱為「抗豬圓環病毒2型的表達載體和轉基因豬及其構建方法」所公布的方法獲得的抗豬圓環病毒2型轉基因克隆豬(f0代)與品種、遺傳背景一致的野生型杜洛克母豬雜交繁育所得。且專利號為201310321355.6,專利名稱為「抗豬圓環病毒2型的表達載體和轉基因豬及其構建方法」所公布的抗豬圓環病毒2型轉基因克隆豬(f0代)包含抗豬圓環病毒2型的sirna、篩選基因neo,綠色螢光蛋白標記基因egfp等外源基因,其中neo與egfp基因融合表達,該f0代抗豬圓環病毒2型轉基因豬來源於本校。
實施例1:利用生物螢光燈觀察f1代抗pcv2轉基因豬的egfp表達。
在藍色光激發條件下,肉眼透過相應的濾光片能觀察到egfp蛋白所呈現的綠色螢光。基於此原理,利用生物螢光燈可以初步鑑定全身組織表達egfp蛋白的f1代抗pcv2轉基因豬。具體操作如下:將轉基因豬和野生型豬擺放在同一處,分別在相同的藍色光背景和白光背景下觀察並拍照。其中,藍光背景下相機拍攝參數為:光圈優先,光圈f2.8,iso4000,手動對焦;白光背景下相機拍攝參數為:光圈優先,光圈f5.6,iso100,手動對焦。同理,對轉基因豬和野生型豬解剖所取的部分器官組織進行觀察並拍照。
檢測結果如附圖1所示,在f1代抗pcv2轉基因豬的口鼻部位以及器官組織腎、肺、心均能觀察到明顯的綠色螢光表達,而陰性對照在同樣條件下未能觀測到綠色螢光。這表明,f1代抗pcv2轉基因豬能穩定遺傳並高效表達整合到f0代轉基因豬基因組的外源基因。
實施例2:利用southernblot檢測外源基因在f1代抗pcv2轉基因豬中的整合。
通過southernblot可以鑑定外源基因在轉基因豬基因組中整合的拷貝數,其原理大致為:先對pegfp-sh2載體序列進行酶切位點分析,選擇一個比較合適的單酶切位點(hindiii),然後用pegfp-sh2質粒設計1拷貝數的對照,用f0代抗pcv2轉基因豬基因組dna作為陽性對照,並以哺乳動物基因組dna大小約為3×109個鹼基對(bp)作為依據,按下述公式計算拷貝對照所需要的質粒量:
質粒量(μg)=插入質粒片段大小(bp)/3*109(bp)×基因組dna量(μg)
從而根據結果估計f1代抗pcv2轉基因豬插入外源基因的拷貝數。具體操作步驟如下:
(1)pcr法製備southernblot所需探針
southernblot所需探針採用pcr標記法製備,其原理為:在表達目的sirna的質粒片段兩端分別設計引物,採用pcrdigprobesynthesiskit來標記,擴增得到的650bppcr產物即為標記好的探針。引物序列及擴增產物長度見表如下:
反應體系的配製如下:
具體操作步驟為:
1)將試劑各組分於冰上解凍,短離心,混勻;
2)按上表中各組分的量由大體積到小體積依次加入反應管中,所有操作均在冰上進行,混勻,短離心;
3)將反應管置於pcr儀中進行擴增反應,程序如下:95℃-2min;95℃-30s,60℃-30s,72℃-40s;30cycles;72℃-7min;
4)檢測探針標記效率:吸取等體積的pcr產物上樣於2%的瓊脂糖凝膠進行電泳,如果可見目的條帶大小正確,並且dig標記探針比未標記對照探針遷移速度稍慢,則表明所標記探針製備成功,-20℃保存備用
(2)southernblot雜交流程
southernblot所用試劑盒為dighighprimednalabelinganddetectionstarterkitii,具體操作步驟根據試劑盒說明書進行。
檢測結果如附圖2所示,表明檢測的6頭f1代抗pcv2轉基因豬存在1-4個整合位點,其中tg3和tg4均只有一個整合位點,而野生型對照豬則沒有獲得southernblot結果,表明本實驗所製備的探針是特異的,f1代抗pcv2轉基因豬特異地整合了目的基因及外源標記基因。
實施例3:利用real-timeqpcr檢測sirna在f1代抗pcv2轉基因豬中的表達。
(1)總rna提取
rna組織樣浸泡於rnalater中並保存於-80℃。應用mirvanamirnaisolationkit提取總rna,具體操作步驟根據說明書進行。
(2)特定sirna的反轉錄
根據目的基因sirna和內參基因mir16序列設計引物,用同一rna模板分別於pcr管中進行反轉錄反應,經引物擴增後得到cdna。引物序列如下:
反轉錄(7.5μl反應體系)mastermix體系的配製如下:
反轉錄mastermix體系的配製過程如下:
i.輕輕混勻,切勿渦旋,離心至管底,然後放置於冰上;
ii.對於每一個7.5μl的反應體系,加入1μl5ng~10ng的總rna;
iii.輕輕混勻,切勿渦旋,短離心至管底,冰上操作;
iv.加入3μl50nm的引物,引物使用前混勻並短離心;
v.混勻,切勿渦旋,短離心至管底,冰上孵育5min;
vi.反轉錄程序為:16℃,30min;42℃,30min;85℃,5min;4℃保存。
(3)特定sirna的real-timeqpcr檢測
i.目的基因sirna及內參基因mir16的qpcr檢測引物及探針如下:
ii.sirnaqpcr20×taqmansirnaassay100μl體系的配製如下:
iii.sirnaqpcr反應體系的配製如下:
上述試劑均在冰上溶解,混勻,短離心至管底。qrcr反應程序為:95℃,10min;95℃,15s,60℃,1min;40cycles。
檢測結果如附圖3所示,結果表明不同f1代轉基因豬之間sirna的表達存在差異,其表達量與southernblot結果中得知的外源目的基因拷貝數多少並不嚴格對應,推測這可能與外源目的基因在基因組上的整合位置有關;而外源基因均為單拷貝的個體tg3的表達量是tg4的4倍左右。
根據實施例2和實施例3的結果表明,tg3轉基因豬個體在外源基因只有一個整合位點,且具有較高的抗pcv2sirna表達量,因而選擇篩選出該tg3個體進行後續研究。
實施例4:利用ipcr(反向pcr)確定外源基因在tg3抗pcv2轉基因豬基因組中的整合位點。
經過southernblot初步判斷外源基因在tg3個體基因組中整合情況後,進一步採用ipcr的方法尋找外源基因的整合位點,一方面可以驗證southernblot的結果,另一方面可以分析外源基因在染色體上的整合位置對轉基因豬的影響,另一方面還可以為刪除標記基因做準備。通過分析southernblot的結果,可以判斷hindiii限制性內切酶同樣適用於ipcr。ipcr具體操作步驟如下:
(1)基因組dna的提取
參照實施例2中的步驟。
(2)基因組dna的酶切及純化
參照實施例2中的步驟,dna用量為2μg。
(3)酶切產物的連接及純化
取上述酶切回收的dna樣品0.2μg~2μg置於0.5ml離心管中,再依次加入連接反應試劑,混勻,短離心,將反應管置於16℃恆溫金屬浴中反應12h-16h,最後純化回收,具體純化步驟如上。
(4)ipcr檢測
ipcr以純化回收的連接產物為模板進行反應,所用引物序列如下:
以上4條引物序列中,其中pb3’treprimary和cmvprimary為一對,用於擴增轉座子3』端和整合位點附近的基因組序列;pb5’treprimary和neoprimary為一對,用於擴增轉座子5』端和整合位點附近的基因組序列。
(5)ipcr產物切膠回收
將ipcr產物全部進行瓊脂糖凝膠電泳,使各條帶儘可能地分開以便切膠回收。切膠回收採用e.z.n.a.gelextractionkit,具體操作步驟根據說明進行。
(6)t-a克隆
經切膠回收的ipcr產物3』末端帶有a鹼基,能直接與instaclonepcrcloningkit中的t-vector進行t-a克隆反應,反應體系如下:
將上述混合液混勻,短離心,至於22℃反應1h,連接後的產物用來轉化dh5α感受態細胞。
(7)轉化
i.將保存於-80℃的感受態細胞取出置於冰上解凍;
ii.將50μl感受態細胞移至滅菌處理的離心管內,加入上述連接產物,輕輕混勻,冰浴30min;
iii.42℃熱擊45s~60s,然後迅速轉移至冰中放置2min,此過程不要搖動離心管;
iv.加入500μl37℃預熱的lb培養基,置於恆溫空氣搖床,37℃,220rpm,復壯1h;
v.取適量復壯後的菌液塗布於經iptg和x-gal處理的la固體培養基,並將其置於37℃至液體被吸收,倒置,37℃培養12h~16h。
(8)藍白斑篩選及菌液pcr鑑定
在上述la培養板上挑取白色單克隆菌落,置於1mlla液體培養基中,37℃,220rpm,培養4h;利用ipcr檢測引物進行菌液pcr鑑定。(9)菌液測序及比對分析
將上述pcr鑑定為陽性的單克隆菌液送往華大基因進行測序,並將成功測序的序列於ncbi上與pegfp-sh2載體序列及豬全基因組序列進行比對,分析尋找整合位點。
檢測結果如附圖4所示,測序及比對結果表明在tg3個體中,外源基因整合在第10號染色體上,位於第10號染色體兩個功能基因cugbpelav-likefamilymember與trans-actingt-cell-specifictranscriptionfactorgata-3之間,不會影響該tg3轉基因豬正常基因的表達,同時外源基因整合在該位點具有較高的表達效果。
實施例5:tg3抗pcv2轉基因豬的耳皮成纖維細胞系的建立。
tg3抗pcv2轉基因豬出生後,經過pcr和southernblot鑑定,確定為後續需要深入研究的個體,採其耳皮組織,在實驗室進行耳皮成纖維細胞建系,方法如下:
(1)用碘酊對要取樣的部位以及耳缺鉗、鑷子、等進行消毒,儘可能地除去耳皮表面的汙垢,剪取一塊小指指甲般大小的組織塊,置於碘酊中泡1min~2min,然後轉移至75%乙醇消毒液進行脫碘並消毒約30s,再用鑷子夾住組織於含2%雙抗的dpbs中涮洗3次,最後快速放入裝有含2%雙抗的無血清dmem的50ml離心管中蓋緊,置於冰盒中儘快帶回實驗室進行下一步的處理,以保證耳皮細胞的活性;
(2)將組織從管中取出置於細胞培養皿中,用含2%雙抗的dpbs洗滌3~5次,剪除毛茬,用手術刀片輕輕刮去表皮細胞,保留真皮層,再將組織泡於碘酊30s,75%乙醇消毒液脫碘30s,含2%雙抗的dpbs涮3~5次,用眼科剪將耳皮組織剪碎成小塊;
(3)用完全培養基清洗組織碎末2次後用1ml的血清重懸,再轉移到新的培養皿中並使其均勻分布,放入37℃、5%co2及飽和溼度的細胞培養箱中,待培養5h~7h至組織小塊貼附於培養皿底後添加完全培養基;
(4)每隔2天換液一次並觀察組織塊周圍細胞爬出情況,待細胞長至80%~90%匯合度時進行傳代培養和凍存。
檢測結果見附圖5,結果表明tg3耳皮成纖維細胞系成功建系的細胞貼壁正常,生長速度良好,呈長梭形或不規則形,富有立體感,有較強的綠色螢光表達。
實施例6:htncre重組酶的表達純化。
(1)將htncre重組酶原核表達質粒轉化於bl21(de3)菌株,37℃培養過夜。在平板上挑單克隆接入3ml的la液體培養基中(含50μg/mlamp),37℃,250rmp振蕩培養至od600=0.5~0.6後,(a)加入0.5mmiptg37℃誘導3h;(b)加入0.1mmiptg16℃誘導過夜;sds-page檢測表達。
(2)重組質粒轉化的bl21(de3)表達菌株接種於100mlla液體培養基中,37℃培養過夜,第二天轉接到1lla液體培養基中,37℃,250rmp振蕩培養至od600=1.0後,加入0.1mmiptg16℃誘導過夜,收菌。
(3)sp-ffcolumn用20mmpb,200mmnacl,1mmedta,ph8.0緩衝液平衡,以20mmpb,1mnacl,1mmedta,ph8.0緩衝液梯度洗脫,收集洗脫組分。
(4)superdex200column以pbs,10%glycerol,ph7.4緩衝液平衡,sp-ffcolumn洗脫收集組分上樣後,以pbs,10%glycerol,ph7.4緩衝液分步洗脫,收集洗脫組分,濃縮得到蛋白產品,檢測蛋白濃度。
檢測結果如附圖6所示,從附圖6中的sds-page凝膠電泳檢測可見,誘導菌株bl21(de3)過量表達產物在分子量大小約為40kda處出現表達量相對較高的明顯條帶,與理論計算的大小42.76kda相符,初步判斷該條帶為目的重組蛋白條帶。本實驗獲得可溶性表達,但表達不明顯,經過大量收集溶菌產物上清,最後從上清中濃縮純化得可溶性htncre重組酶1.2mg,濃度為0.6mg/ml,儲存於pbs、10%glycerol、ph7.4緩衝液中,小管分裝後於-80℃長期保存備用,儘量避免反覆凍融。
實施例7:利用htncre刪除tg3個體耳皮成纖維細胞系neo/egfp外源標記基因。
(1)復甦tg3個體耳皮成纖維細胞於60mm細胞培養皿,長滿後傳代至6孔板,培養條件為37℃,5%co2。
(2)待細胞密度達80%左右時,用dpbs洗2次,加入opti-mem稀釋的htncre,使其終濃度為200ng/μl,孵育3h後吸走htncre工作液,用dpbs洗2次,加入10%fbs的完全培養基(含1%雙抗)繼續培養。
(3)24h後,用0.25%的胰酶消化細胞,用細胞計數儀對細胞進行計數,按200個細胞/皿傳至100mm細胞培養皿中,每隔3天換一次液並觀察細胞單克隆的形成情況。
(4)當細胞培養至10天左右,細胞克隆團細胞數達到200個左右時,用marker筆在倒置顯微鏡的觀察下於細胞培養皿底部圈出細胞克隆團所在位置,並在克隆團外圍再圈出無雜細胞的安全區域,同時用螢光顯微鏡觀察細胞克隆團有無螢光,做好標記,以確保挑取更純的無螢光細胞單克隆。
(5)吸走完全培養基,用dpbs洗2次,用鑷子將克隆環小心準確放在已做標記的細胞克隆上,每個克隆環內加入兩滴0.25%胰酶,在顯微鏡下觀察至細胞變圓時,加入50μl左右的fbs終止消化,用移液器小心吹起細胞並轉移至24孔板中,按24孔-6孔-60mm的順序進行傳代培養,可在傳代過程中一分為二,一份用於pcr鑑定,一份凍存備用。每傳一代都需進行螢光觀察,進一步確保篩選到的細胞單克隆中無未刪除螢光標記的雜細胞。
檢測結果用實施例6中表達純化的htncre重組酶處理f1代抗pcv2轉基因豬tg3耳皮成纖維細胞系,由於綠色螢光蛋白在htncre重組酶處理前就已經存在,在htncre刪除標記之後其代謝需要一定的時間,所以細胞系標記被刪除的效果在24h內並不能明顯觀察到,細胞始終有可見的綠色螢光。但隨著細胞的繼續分裂生長,綠色螢光越來越弱。如附圖7所示的是,tg3細胞系在htncre處理裡後第7天進行流式細胞分析,發現tg3表達綠色螢光的細胞由標記未刪除前的99.8%變為刪除標記後的4.8%,標記刪除效率高達95%。
檢測結果如附圖8所示:htncre處理f1代抗pcv2轉基因豬tg3耳皮成纖維細胞系後,稀釋培養並用克隆環進行細胞單克隆篩選,發現未經過htncre刪除標記的細胞單克隆表達較強的綠色螢光(40×),而經過htncre刪除標記的則未能觀察到有綠色螢光表達。
同時,由於標記基因neo與綠色螢光蛋白基因egfp為融合表達,且位於兩個loxp之間,因而egfp被刪除的同時,neo基因也會被同時刪除。
在其他實施例中,htncre重組酶濃度還可以為100ng/μl,150ng/μl或300ng/μl等濃度,均可以具有較高效率的刪除外源標記基因的效果,只是優選的採用200ng/μl時,刪除效果最好。
實施例8:利用pcr檢測刪除標記基因的細胞單克隆。
(1)細胞dna提取參照實施例2中的步驟;
(2)利用pcr檢測neo/egfp融合表達標記基因和兩個loxp之間的序列,所用引物如下:
用pcr對篩選到的f1代抗pcv2轉基因豬tg3細胞單克隆的neo/egfp標記以及兩個loxp之間的標記序列進行檢測,如附圖9所示,篩選到的c1~c7無綠色螢光細胞單克隆均未檢測到neo/egfp標記以及loxp之間的標記序列,而陽性對照中的質粒、未處理以及處理後的混合細胞中則均能檢測到,說明篩選到的細胞單克隆中的標記已被htncre重組酶介導的刪除反應刪除掉,其刪除反應原理以及刪除標記後的pcr產物測序驗證見附圖10,即標記刪除前能擴增到980bp的neo/egfp標記以及3211bp的兩個loxp之間的序列,而標記刪除後擴增不到980bp的neo/egfp標記以及只能擴增到530bp兩個loxp之間序列被刪除後剩下的片段。
實施例9:利用scnt製備無neo/egfp標記基因的抗pcv2轉基因克隆豬。
將實施例7中獲得的無neo/egfp標記基因的tg3個體耳皮成纖維細胞系作為核供體細胞,注射到卵母細胞中,構建重構胚胎進行體細胞克隆(scnt),並採用常規育種技術進行培育,即可獲得抗pcv2的轉基因豬。實施例10:利用生物螢光燈檢測無neo/egfp標記基因抗pcv2轉基因克隆豬的egfp表達。
檢測方法同實施例1。
用生物螢光燈對獲得的無neo/egfp標記基因抗pcv2轉基因克隆豬進行egfp表達檢測,選擇大小相似的表達egfp的豬作為對照,對照豬可見較明顯的egfp表達,尤其是在口蹄部,而無neo/egfp標記抗pcv2轉基因克隆豬則觀察不到egfp的表達,如附圖11,判斷通過實施例9獲得的無neo/egfp標記抗pcv2轉基因克隆豬其egfp標記被刪除,由於neo基因與egfp基因融合表達,egfp基因不表達時,neo基因也不會表達。
實施例11:利用pcr檢測無neo/egfp標記基因的抗pcv2轉基因克隆豬。
檢測方法同實施例8。經pcr進一步檢測,檢測結果如附圖12,結果表明,實施例9中獲得的抗pcv2轉基因小豬均成功刪除neo/egfp外源標記基因,而目的基因抗pcv2sirna基因序列予以保留,也進一步證明篩選用於製備無標記抗pcv2轉基因克隆豬的細胞單克隆較純且成功刪除了標記基因,與綠色螢光檢測結果相符合。表明通過上述方法獲得了無外源neo/egfp標記基因的抗pcv2轉基因克隆豬。
以上所述的僅是本發明的一些實施方式。對於本領域的普通技術人員來說,在不脫離本發明創造構思的前提下,還可以做出若干變形和改進,這些都屬於發明的保護範圍。