量子點合成裝置及量子點合成方法與流程
2023-05-10 04:56:27 4
本發明涉及量子點合成領域,具體涉及一種量子點合成裝置及通過該量子點合成裝置實施的量子點合成方法。
背景技術:
現有技術中,量子點的合成方法為溶液法工藝,主要由圍繞著加熱套的燒瓶、能使燒瓶內部溶液均勻的磁力攪拌器、以及控制溶液溫度的溫度調節器和溫度計組成。另外,添加了維持穩定濃度的電容器和能轉換真空/氮氣氛圍的多支管組成。
合成方法為利用前驅體(precursor)的有機化合物合成核(core),為了使合成的核和穩定劑在混合攪拌的反應器中形成適當的殼,反覆注入前驅體形成結構穩定的核/殼結構,最近還開發出了one-pot工藝,將所有反應物質一次性放入反應器中再添加殼工序的方法。
如此,溶液工藝合成方法變成大容量化時,為了反應物質內的均勻性,有時也會用葉輪代替磁力攪拌器,但是因為不均勻的溫差、不均勻的環境導致反應物內濃度的差異以及組合的差異。量子點合成是奧斯特瓦爾德熟化(ostwaldripening),也是溫度和時間的函數,據悉此技術是小的粒子與相對大的顆粒合體之後生長,當形成所需大小的納米粒子時停止粒子生長的技術。但是目前的方法是通過冷卻控制粒子生長時,會產生溫度差,很難合成出大小均勻的粒子。因此,這些因素還會影響量子點納米粒子的發光特性和散布大小均勻的粒子。
為了得到一定大小的納米粒子,進行急速冷卻。此反應物中也存有反應的副產物、有機物,所以通過離心過濾除去有機物。納米粒子分離可通過丙酮或乙醇等強極性溶劑,利用離心過濾區分粒子的大小。各個反應過程中通過磁棒或攪拌機進行強制攪拌,雖然這是眾所周知能達到比較均勻攪拌的方法,但是根據反應物的量,反應攪拌速度會有所不同,這會影響粒子的大小和穩定納米粒子的形成。
這種現有的合成方法還另外需要前驅體反應器、核反應器、核/殼反應器等,因為是在各自反應後重新進行計量、測量,然後進行合成,所以存在無法連續作業的缺點。而且因為反應時間、粒子分離等差異,有可能無法得到具有均勻的特點的納米粒子。
技術實現要素:
為了解決上述問題,本發明公開了一種量子點合成裝置。
本發明還公開了一種通過該量子點合成裝置實施的量子點合成方法。
本發明為實現上述目的所採用的技術方案是:
一種量子點合成裝置,其包括依次連接的前驅體反應區、核反應區、核/殼反應的核/殼反應區和保管容器,上述各反應區分別包括至少一個螺旋狀反應管,上述螺旋狀反應管內壁上設有與該螺旋狀相反方向的使注入反應管中的反應物因摩擦產生冒泡效應的途徑槽,上述各反應區的前段包覆有加熱區,上述各反應區的後段包覆有冷卻區,於上述各反應區的前端分別設有若干個注入口,於該核反應區的後端、核/殼反應區的後端分別設有若干個浮遊物排出口,所述各反應區的前端連接有加壓裝置。
所述加熱區、冷卻區分別包括纏繞於該螺旋狀反應管上的溫度控制管,所述溫度控制管中填充滿用於控制溫度的液態水或油,該加熱區、冷卻區的溫度控制管分別與加熱裝置、外部冷卻器連接。
所述核反應區的後段、核/殼反應區的後段均設有急速旋轉區域和浮遊物排出區域,所述浮遊物排出口設於該浮遊物排出區域。
該急速旋轉區域中螺旋狀反應管的管間距離小於核反應區、核/殼反應區前段的螺旋狀反應管的管間距離,該浮遊物排出區域的管間距離大於核反應區、核/殼反應區前段的螺旋狀反應管的管間距離。
該急速旋轉區域的直徑為核反應區、核/殼反應區前段的螺旋狀反應管直徑的三倍;該急速旋轉區域的彎曲度比核/殼反應區前段的螺旋狀反應管減少50%。
一種通過上述量子點合成裝置實施的量子點合成方法,其包括以下步驟:
(1)前驅體生成原料於前驅體反應區反應形成前驅體;
(2)部分前驅體於核反應區反應形成核;
(3)部分前驅體與核於核/殼反應區反應形成核/殼結構,即形成量子點;
(4)量子點輸送至保管容器進行保存。
所述步驟(1)中,將前驅體反應區的螺旋狀反應管進行真空排氣後轉換為氮氣環境,將前驅體生成原料通過注入口注入前驅體反應區的螺旋狀反應管,通過加熱區將螺旋狀反應管加熱到設定溫度,利用加壓裝置通過氮氣壓力將前驅體生成原料經過螺旋狀反應管進行混合、反應,根據反應所需時間調整螺旋狀反應管的長度,根據攪拌所需時間調整螺旋狀反應管的內徑或長度,反應完成後生產的前驅體轉移至冷卻區進行冷卻;
依次類推,分別製得cd前驅體、s前驅體、zn前驅體和se前驅體。
所述步驟(2)中,將核反應區螺旋狀反應管進行真空排氣後轉換為氮氣環境,將cd前驅體、氯化三辛基膦、1-十八碳烯通過注入口注入核反應區的螺旋狀反應管進行混合,通過加熱區將螺旋狀反應管加熱到設定溫度,當反應物經過設定時間及通過設定長度後,加入油胺和se前驅體,從而生成cdse核,反應生成的雜質通過急速旋轉區域加速後會浮在反應物上,於浮遊物排出區域的浮遊物排出口將雜質排出,之後獲得精製的cdse核。
所述步驟(3)中,將核/殼反應區進行真空排氣後轉換為氮氣環境,將cdse核、1-十八碳烯和油胺注入核/殼反應區的螺旋狀反應管,通過加熱區將螺旋狀反應管加熱到設定溫度,使cdse核、1-十八碳烯和油胺反應,按間隔時間依次加入cd前驅體與zn前驅體,s前驅體、zn前驅體與cd前驅體,s前驅體、zn前驅體與cd前驅體,和zn前驅體與s前驅體,從而合成cdse/cdzns/zns量子點。
所述步驟(3)中,反應生成的雜質通過急速旋轉區域加速後會浮在反應物上,於浮遊物排出區域的浮遊物排出口將雜質排出,獲得完成精製的cdse/cdzns/zns的核殼納米粒子。
本發明的有益效果為:本發明的量子點合成裝置能提高量子點的穩定性及粒子大小的均勻,同時能使其沒有缺陷,因此能使量子效率最大化,從而合成出提升了發光效率及鮮明度的量子點。
下面結合附圖與具體實施方式,對本發明進一步說明。
附圖說明
圖1是本發明的結構示意圖;
圖2是本發明中前驅體反應區的結構示意圖;
圖3是本發明中核反應區的結構示意圖;
圖4是本發明中核/殼反應區的結構示意圖;
圖5是uvlight照射的量子點圖片;
圖6是本發明製得的量子點發光特性圖;
圖7是本發明製得的量子點吸光特性圖。
具體實施方式
實施例,參見圖1至圖7,本實施例公開了一種量子點合成裝置,其包括依次連接的前驅體反應區1、核反應區2、核/殼反應的核/殼反應區3和保管容器4,上述各反應區分別包括至少一個螺旋狀反應管,上述螺旋狀反應管內壁上設有與該螺旋狀相反方向的使注入反應管中的反應物因摩擦產生冒泡效應的途徑槽,上述各反應區的前段包覆有加熱區,上述各反應區的後段包覆有冷卻區,於上述各反應區的前端分別設有若干個注入口,於該核反應區2的後端、核/殼反應區3的後端分別設有若干個浮遊物排出口,所述各反應區的前端連接有加壓裝置。
所述加熱區、冷卻區分別包括纏繞於該螺旋狀反應管上的溫度控制管,所述溫度控制管中填充滿用於控制溫度的液態水或油,該加熱區、冷卻區的溫度控制管分別與加熱裝置、外部冷卻器連接,能給各個反應區提供恆定溫度。
當反應物通過螺旋狀反應管的時候會因摩擦產生冒泡效應,因此會比磁棒或攪拌機更加穩定均勻的攪拌,而反應時間可以根據螺旋狀反應管的長度調整反應時間,從而減少量子點合成時不必要的反應時間。此外,還可以變換螺旋狀反應管的直徑,調整通過螺旋狀反應管的反應物的流速,變更合成條件。螺旋狀反應管的外部填充著可以調節溫度的液狀水或者油,所以螺旋狀反應管的起始點和螺旋狀反應管的末端的合成條件會一致。也就是說,隨著螺旋狀反應管外填充著的加熱、冷卻媒介能分段控制,使其變得可行。
所述核反應區2的後段、核/殼反應區3的後段均設有急速旋轉區域和浮遊物排出區域,所述浮遊物排出口設於該浮遊物排出區域。
該急速旋轉區域中螺旋狀反應管的管間距離小於核反應區2、核/殼反應區3前段的螺旋狀反應管的管間距離,該浮遊物排出區域的管間距離大於核反應區2、核/殼反應區3前段的螺旋狀反應管的管間距離。
該急速旋轉區域的直徑為核反應區2、核/殼反應區3前段的螺旋狀反應管直徑的三倍;該急速旋轉區域的彎曲度比核/殼反應區3前段的螺旋狀反應管減少50%。
而且washing和粒子分離工藝,因螺旋狀反應管的彎曲和直徑的變化變為可能。當通過彎曲度大直徑小的區間時,在通過的起始點注入一定量的乙醇或丙酮,會產生反應蒸氣壓,同時會讓反應物出現冒泡效應,為了控制流速將直徑擴大三倍、彎曲度減少50%的話,能得到與離心過濾相同的效果。此時,通過路徑中上、下分離的區間,去除上端的有機物,然後讓下端的反應物,即核步入下一個工序。必要時,為了流動的流暢,可以使用己烷(hexane)等溶劑。
核/殼工藝的優點是,用於積累殼的注入口位於螺旋狀反應管的一定長度上,所以能始終積累大小均勻的殼,積累第二層殼、第三層殼時,可以根據計算的核大小和積累的大小控制通過螺旋狀反應管積累殼體的cd、zn、s等的注入口距離,因此能始終積累均勻且沒有缺陷的殼體。
一種通過上述量子點合成裝置實施的量子點合成方法,其包括以下步驟:
(1)前驅體生成原料於前驅體反應區1反應形成前驅體;
(2)部分前驅體於核反應區2反應形成核;
(3)部分前驅體與核於核/殼反應區3反應形成核/殼結構,即形成量子點;
(4)量子點輸送至保管容器4進行保存。
所述步驟(1)中,將前驅體反應區1的螺旋狀反應管進行真空排氣後轉換為氮氣環境,將前驅體生成原料通過注入口注入前驅體反應區1的螺旋狀反應管,通過加熱區將螺旋狀反應管加熱到設定溫度,利用加壓裝置通過氮氣壓力將前驅體生成原料經過螺旋狀反應管進行混合、反應,根據反應所需時間調整螺旋狀反應管的長度,根據攪拌所需時間調整螺旋狀反應管的內徑或長度,反應完成後生產的前驅體轉移至冷卻區進行冷卻;
依次類推,分別製得cd前驅體、s前驅體、zn前驅體和se前驅體。
所述步驟(2)中,將核反應區2螺旋狀反應管進行真空排氣後轉換為氮氣環境,將cd前驅體、氯化三辛基膦、1-十八碳烯通過注入口注入核反應區2的螺旋狀反應管進行混合,通過加熱區將螺旋狀反應管加熱到設定溫度,當反應物經過設定時間及通過設定長度後,加入油胺和se前驅體,從而生成cdse核,反應生成的雜質通過急速旋轉區域加速後會浮在反應物上,於浮遊物排出區域的浮遊物排出口將雜質排出,之後獲得精製的cdse核。
所述步驟(3)中,將核/殼反應區3進行真空排氣後轉換為氮氣環境,將cdse核、1-十八碳烯和油胺注入核/殼反應區3的螺旋狀反應管,通過加熱區將螺旋狀反應管加熱到設定溫度,使cdse核、1-十八碳烯和油胺反應,按間隔時間依次加入cd前驅體與zn前驅體,s前驅體、zn前驅體與cd前驅體,s前驅體、zn前驅體與cd前驅體,和zn前驅體與s前驅體,從而合成cdse/cdzns/zns量子點。
所述步驟(3)中,反應生成的雜質通過急速旋轉區域加速後會浮在反應物上,於浮遊物排出區域的浮遊物排出口將雜質排出,獲得完成精製的cdse/cdzns/zns的核殼納米粒子。
本發明的量子點合成裝置能提高量子點的穩定性及粒子大小的均勻,同時能使其沒有缺陷,因此能使量子效率最大化,從而合成出提升了發光效率及鮮明度的量子點。
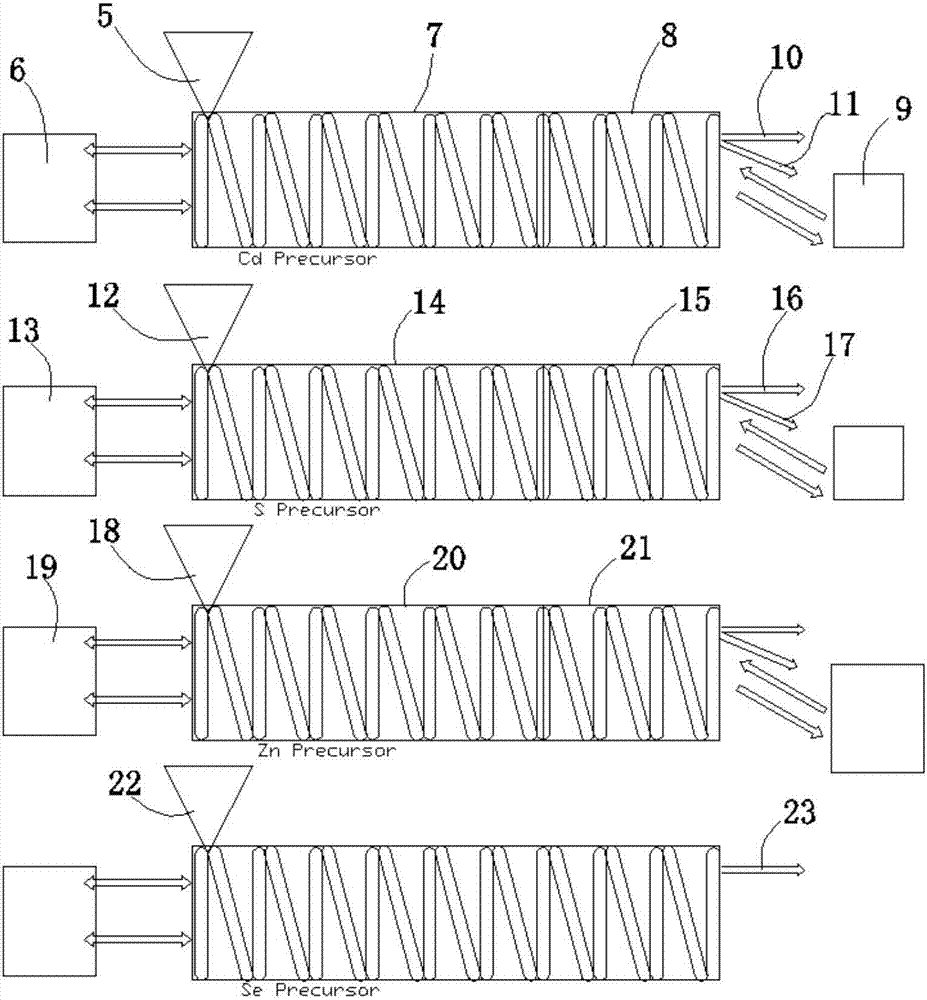
具體的,在前驅體合成的過程中,如圖2所示,圖2中為了生成各種前驅體共設有多個螺旋狀反應管。為了製作cd(鎘)前驅體,在注入口5以0.05~1.0左右的摩爾濃度放入oleicacid和cdoxide,真空排氣後轉換成氮氣環境。螺旋狀反應管的溫度則是通過加熱裝置6加熱成170攝氏度的液體,不斷循環,將液狀媒介加熱為一定的溫度,然後在加熱區7的螺旋狀反應管外部循環。
當溫度達到170攝氏度恆溫時,利用氮氣壓力使通過注入口5注入的原料經過螺旋狀反應管與反應物混合,反應所需的時間根據螺旋狀反應管的長度決定。通過螺旋狀反應管合成的前驅體和核以及核/殼工序的反應時間可能比傳統攪拌合成時的反應時間要短,攪拌時間可根據螺旋狀反應管內徑的變化和長度的變化進行調節。完成反應的cd前驅體會轉移到冷卻區8內,此時,通過外部冷卻器9供應的冷卻水進行冷卻,最適合的冷卻水溫度為50攝氏度~60攝氏度。完成冷卻的cd前驅體與核合成時通過管道10輸送,與核/殼合成時通過管道11輸送。進行所有反應時,反應物的移送通過氮氣強制進行。氮氣壓力會根據螺旋狀反應管的內徑和反應時間隨時改變。
為了製作s(硫)的前驅體,在注入口12計量注入了0.1m的1-十八碳烯(octadecene)和s粉末,真空排氣後轉換為氮氣環境。加熱裝置13使加熱區14的溫度恆定在120攝氏度在120攝氏度,反應物在設定時間經過加熱區14處的螺旋狀反應管。完成合成的s前驅體在冷卻區15進行冷卻,然後與cd前驅體一樣,利用氮氣壓力進行移送,為了核合成通過管道16輸送,為了合成核/殼通過管道17輸送。
為了合成zn(鋅)前驅體,在注入口18中計量注入摩爾濃度為0.5~1.5m的1-油酸(octadecene)和十八烯(oleicacid)以及氧化鋅(znoxide),真空排氣後轉換成氮氣環境。螺旋狀反應管的溫度經過加熱裝置19維持在300攝氏度,反應物在加熱區20合成。完成合成的反應物通過注入到管內的氮氣壓力移送到冷卻區21,冷卻區21的溫度控制在100~120攝氏度。這些cd、s、zn的前驅體在常溫冷卻時,會出現凝固、凝膠狀或固體化,所以為了順暢的進行反應,應保持一定溫度。
為了合成se(硒),在注入口22計量注入摩爾濃度為0.7~1.5mm的三辛基(trioctylphosphine)和硒粉,真空排氣後轉換成氮氣環境。反應管的溫度在加熱區維持為50~60攝氏度,為了合成核通過管道23輸送。
如圖3所示,在核的合成過程中,在注入口24中計量注入氯化三辛基膦(trioctylphosphineoxide)和1-十八碳烯(octadecene),在注入口25中再計量注入油胺(oleylamine)和se前驅體,真空排氣後轉換為氮氣環境。cd前驅體通過注入口24被注入到螺旋狀反應管中,與通過注入口25注入的反應物進行混合。通過加熱區26加熱到270~290攝氏度。為核合成的螺旋狀反應管推薦長度為250㎝,經過3分20秒之後,在通過注入口24注入的反應物通過注入口25之前,通過注入口25注入油胺(oleylamine)和se前驅體。從而混合注入的反應物,隨之核會增長。核的大小決定發光波長,核的大小是由螺旋狀反應管的長度和反應物的通過時間決定。
完成反應的反應物中不僅有cdse核,還混合著很多有機物或反應副產物等許多雜質,為了減少結晶攝氏度和缺陷,必須完全清除雜質。為此,要對增長的核進行急速冷卻做成希望的大小,並且為了分離核和雜質,應把反應物移送到急速旋轉區域27。為了分離cdse核和雜質,在注入口28注入極性強的乙醇、丙酮等,變化螺旋狀反應管的流速即可解決。冷卻區29溫度維持到0~4攝氏度,通過急速旋轉區域27的反應物會以非常快的流速經過,隨著螺旋狀反應管的弧度變得柔和,比較輕的有機物等雜質會浮遊在反應物上流過。此時,通過浮遊物排出口30、31、32可以將浮遊物清除到外部,也就是利用維持壓力比螺旋狀反應管壓力稍微低一點的電容器,通過浮遊物排出口30、31、32清除浮遊的雜質。完成精製的cdse核為了殼工序被通過管道33輸送。當然,在核反應中也將利用氮氣壓力移送反應物。
如圖4所示,螺旋狀反應管上各個注入口之間的距離相等,在核/殼(core/shell)的合成過程中,在注入口40中計量注入cdse核、1-十八碳烯(octadecene)和油胺(oleylamine),真空排氣後轉換為氮氣環境。發光波長是由核的波長和殼的厚度所決定。因為粒子的大小和發光波長具有關聯。加熱區41的溫度控制為220220~250攝氏度,達到240攝氏度通過注入口40注入的反應物會因氮氣壓力隨著螺旋狀反應管進行加熱反應。總反應時間會根據殼的層數有所不同。殼的層數最好為偶數,為了積累殼體,在注入口41中注入cd前驅體和zn前驅體。這些將通過螺旋狀反應管以氮氣壓力進行注入,所以不需要額外的真空排氣。為了再積累第二層殼體,在注入口42中注入cd前驅體、zn前驅體和s前驅體,為了積累第三層殼體,在注入口43中注入cd前驅體、zn前驅體和s前驅體,為了積累第四層殼體,在注入口44注入zn前驅體和s前驅體,然後進行反應。第四層殼體為最外圍的殼,積累zns殼能改善發光特性。如此進行反應的話,就能合成cdse/cdzns/zns的核/殼結構量子點。與核相同,完成反應的反應物中不僅有cdse/cdzns/zns量子點,還混合有很多有機物或反應副產物等雜質,為了減少結晶攝氏度和缺陷必須完全清除雜質。為了分離合成的納米粒子和雜質,應將反應物移送到急速旋轉區域45。與核相同,為了分離合成的納米粒子和雜質,往注入口46注入極性強的乙醇、丙酮等,變化螺旋狀反應管的流速即可解決。冷卻區47溫度維持到0~4攝氏度,通過螺旋狀反應管的反應物會以非常快的流速經過,隨著螺旋狀反應管的弧度變得柔和,比較輕的有機物等雜質會浮遊在反應物上流過。此時,通過浮遊物排出口48、49、50可以將浮遊物清除到外部,也就是利用維持壓力比反應管壓力稍微低一點的電容器,通過浮遊物排出口48、49、50清除浮遊的雜質。完成精製的cdse/cdzns/zns的核殼納米粒子會排放到保管容器4中。納米粒子的大小,及發光波長會根據核的波長、殼的數量而定,為了使納米粒子變大,會積累相對較多的殼,這樣會降低發光特性,因此推薦的殼體層數為4~6層。
例如,在510nm的核上積累四層是會變成525nm,積累六層會變成530nm。而且在600nm的核上積累四層時會變成620nm,積累六層會變成625nm。利用這樣的關係,可以根據核的大小和殼的層數制定最終想取得的量子點納米粒子的發光波長。
如上述,可以通過螺旋狀反應管,得到穩定且均勻、不含雜質、沒有缺陷、發光特性優秀的量子點納米粒子。
如圖5所示,其為通過本發明方法合成的量子點納米粒子,通過uvlight照射的量子點圖片。
如圖6所示,該量子點納米粒子的發光特性如下:x軸為發光波長(nm),y軸是絕對強攝氏度。發光特性的測定可利用horibafluoromax-4spectrofluorometer,激發波長為385nm。
如圖7所示,該量子點納米粒子的吸收特性如下:x軸為吸收波長(nm),y軸是絕對強攝氏度。通過agilentcary300spectrophotometer測定了吸光特性。
以上所述,僅是本發明的較佳實施例而已,並非對本發明作任何形式上的限制。任何熟悉本領域的技術人員,在不脫離本發明技術方案範圍情況下,都可利用上述揭示的技術手段和技術內容對本發明技術方案做出許多可能的變動和修飾,或修改為等同變化的等效實施例。故凡是未脫離本發明技術方案的內容,依據本發明之形狀、構造及原理所作的等效變化,均應涵蓋於本發明的保護範圍內。