雷米普利分子印跡整體柱及其製備方法與應用與流程
2023-05-10 09:42:17
本發明屬於高效液相色譜整體柱技術領域,具體涉及一種雷米普利分子印跡整體柱及其製備方法與應用。
(二)
背景技術:
分子印跡技術起源於Pauling的抗體理論,是一種對特定模板分子及其結構類似物具有特異性識別作用的印跡聚合物的製備和分析應用的新興技術。此技術具有選擇性好、穩定性高、使用壽命長和應用範圍廣等優點,已被廣泛應用於樣品前處理(Xu Z,Hu Y,Hu Y,et al.Investigation of ractopamine molecularly imprinted stir bar sorptive extraction and its application for trace analysis of beta2-agonists in complex samples[J].Journal of Chromatography A,2010,1217(22):3612-3618.)、色譜分離(Zhang H,Ye L,Mosbach K.Non-covalent molecular imprinting with emphasis on its application in separation and drug development[J].Journal of Molecular Recognition,2006,19(4):248–259.)、手性藥物的拆分(Cormack P A G,Mosbach K.Molecular imprinting:recent developments and the road ahead[J].Reactive&Functional Polymers,1999,41(s 1–3):115-124.)、固相萃取(Pichon V.Selective sample treatment using molecularly imprinted polymers.[J].Journal of Chromatography A,2007,1152(1-2):41–53.c)等諸多領域。
傳統的分子印跡聚合物填料採用本體聚合的方法,過程繁瑣、複雜、費時。而整體柱是一種將功能單體、致孔劑、交聯劑和引發劑等混合物在管狀柱內通過原位聚合製備形成的連續床固定相,具有製備簡單快捷、結構均勻穩定、滲透性好、易修飾等優點,被譽為第四代色譜固定相。分子印跡整體柱將分子印跡技術和整體柱技術有機結合,不僅解決了分子印跡聚合物製備繁瑣費時的問題,而且提高了整體柱的選擇性,有效提高了液相色譜的分離效能,已被應用於醫藥、生物和環境等領域,是目前分析化學領域的研究熱點。
(三)
技術實現要素:
本發明的目的是提供一種雷米普利分子印跡整體柱及其製備方法,以及在高效液相色譜分離雷米普利和普利類混合物中的應用,本發明製備方法具有簡單、快捷、易操作、易控制等優點。雷米普利化學名稱:N-[1(S)-羰乙氧基-3-苯基-丙基]-(S)-丙氨醯基-順橋-2-氮雜二環[3,3,0]辛烷-3(S)-羧酸,化學結構式:
為實現上述目的,本發明採用如下技術方案:
一種雷米普利分子印跡整體柱,其製備方法為:
將雷米普利(模板分子)、功能單體加入致孔劑和十二醇(溶劑)的混合液中,室溫(20~30℃)超聲處理20~45min,再加入乙二醇二甲基丙烯酸酯(EDMA,交聯劑)、偶氮二異丁腈(AIBN,引發劑),繼續於室溫下超聲處理20~45min至物料溶解,之後通入惰性氣體以除去溶解氧,得到反應混合物;將所得反應混合物加到空柱管中,密封柱管兩端,置於45~65℃(優選55℃)下進行熱引發聚合反應12~36h(優選18h),製得整體柱,之後經柱衝洗,即得成品。
所述的功能單體為甲基丙烯酸、甲基丙烯酸丁酯、丙烯醯胺、甲基丙烯酸縮水甘油酯、苯乙烯中的一種或任意兩種以任意比例的組合,優選甲基丙烯酸。
所述的致孔劑為N,N-二甲基甲醯胺、甲苯或環已醇;
所述致孔劑和十二醇的混合液中,致孔劑與十二醇的體積比為1:1.57~6(優選1:4.55);
所述致孔劑和十二醇的混合液的體積用量為功能單體和乙二醇二甲基丙烯酸酯體積之和的1.74~4.26倍(優選2.68~3.23倍)。
所述的乙二醇二甲基丙烯酸酯與功能單體的體積比為2.1~9:1(優選3.9~5.5:1)。
所述的雷米普利的質量用量以功能單體的體積計為0.24~1.20g/ml(優選0.72~0.95g/ml)。
所述的偶氮二異丁腈的質量用量以功能單體的體積計為0.07~0.28g/ml(優選0.14~0.21g/ml)。
通常,所述的惰性氣體為氮氣,流速為10ml/L,通氣時間5~30min。
所述柱衝洗的方法為:聚合反應完成後,將製得的整體柱依次用甲醇、甲醇-醋酸(體積比8~4:1)混合液和水分別衝洗,以洗去模板分子、致孔劑等,最後再用甲醇-水(體積比10:90~90:10)混合液充分衝洗至基線平穩,即得所述的雷米普利分子印跡整體柱。
更進一步,所述柱衝洗的方法為:
聚合反應完成後,將製得的整體柱連接到高效液相色譜儀上,依次用0.1mL/min甲醇衝洗0.5h、1mL/min甲醇衝洗1h、1mL/min甲醇與醋酸體積比8~4:1的混合液衝洗1h、1mL/min水衝洗1h,以洗去模板分子、致孔劑和溶劑,最後再用甲醇-水(體積比10:90~90:10)混合液衝洗整體柱至基線平穩,即得到所述的雷米普利分子印跡整體柱。
根據本發明方法製備得到的雷米普利分子印跡整體柱可應用於高效液相色譜分離,所述高效液相色譜柱分離模式為反相模式,分離物質為普利類化合物的混合物,所述的普利類化合物例如:卡託普利、雷米普利、苯那普利、喹那普利等。
本發明所述雷米普利分子印跡整體柱的製備方法簡單、快捷、易操作、易控制,按照本發明方法製備的雷米普利分子印跡整體柱,可簡便、快捷地對普利類混合物進行分離,具有很好的應用前景。
(四)附圖說明
圖1a為實施例1中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖1b為實施例1中普利類混合試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖2a為實施例2中雷米普利試樣在33mg模板分子的整體柱中反相模式下色譜圖;
圖2b為實施例2中普利類混合試樣在33mg模板分子的整體柱中反相模式下色譜圖;
圖3a為實施例3中雷米普利試樣在167mg模板分子的整體柱中反相模式下色譜圖;
圖3b為實施例3中普利類混合試樣在167mg模板分子的整體柱中反相模式下色譜圖;
圖4為對比實施例4中普利類混合試樣在非印跡整體柱中反相模式下色譜圖;
圖5為實施例5中雷米普利試樣在166.6mg模板分子的整體柱中反相模式下色譜圖;
圖6為實施例6中雷米普利試樣在33.3mg模板分子的整體柱中反相模式下色譜圖;
圖7為實施例7中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖8為實施例8中雷米普利試樣在167mg模板分子的整體柱中反相模式下色譜圖;
圖9為實施例9中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖10為實施例10中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖11a為實施例11中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖11b為實施例11中普利類混合試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖12a為實施例12中雷米普利試樣在33mg模板分子的整體柱中反相模式下色譜圖;
圖12b為實施例12中普利類混合試樣在33mg模板分子的整體柱中反相模式下色譜圖;
圖13a為實施例13中雷米普利試樣在133mg模板分子的整體柱中反相模式下色譜圖;
圖13b為實施例13中普利類混合試樣在133mg模板分子的整體柱中反相模式下色譜圖;
圖14為實施例14中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖15為實施例15中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖;
圖16為實施例16中雷米普利試樣在100mg模板分子的整體柱中反相模式下色譜圖。
(五)具體實施方式
下面結合具體實施例對本發明進行進一步闡述,但所述實施例並不限制本發明的保護範圍。
實施例1
整體柱製備:
將100mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和溶劑,最後再用甲醇水溶液(體積比50:50)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
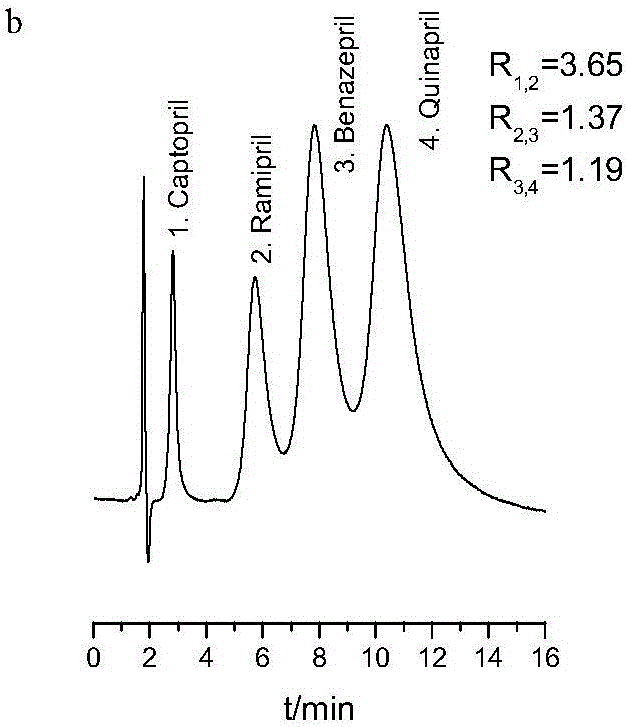
以實施例1製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下對雷米普利(1.0mg/ml)試樣溶液及雷米普利(1.5mg/ml)、苯那普利(3.0mg/ml)、喹那普利(3.0mg/ml)和卡託普利(0.5mg/ml)混合試樣進行分離(用於配製試樣的溶劑為水,以下實施例均同)。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1.0ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,成功分離雷米普利試樣及四種普利類藥物混合試樣。所得色譜圖如圖1a和1b所示,混合試樣中各物質的出峰順序依次為:卡託普利、雷米普利、苯那普利、喹那普利,卡託普利與雷米普利、雷米普利與苯那普利以及苯那普利與喹那普利之間的分離度分別為3.65、1.37、1.19,具有較好的分離效果。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為4000。
實施例2
整體柱製備:
將33mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和溶劑,最後再用甲醇水溶液(體積比50:50)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例2製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下對雷米普利(1.0mg/ml)試樣溶液及雷米普利(1.5mg/ml)、苯那普利(3.0mg/ml)、喹那普利(3.0mg/ml)和卡託普利(0.5mg/ml)混合試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH2.5)及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1.0ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,成功分離雷米普利試樣及四種普利類藥物混合試樣。所得色譜圖如圖2a和2b所示,混合試樣中各物質的出峰順序依次為:卡託普利、雷米普利、苯那普利、喹那普利,卡託普利與雷米普利、雷米普利與苯那普利以及苯那普利與喹那普利之間的分離度分別為2.95、1.14、1.09,具有較好的分離效果。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為2773。
實施例3
整體柱製備:
將167mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和溶劑,最後再用甲醇水溶液(體積比50:50)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例3製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下對雷米普利(1.0mg/ml)試樣溶液及雷米普利(1.0mg/ml)和喹那普利(1.0mg/ml)混合試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1.0ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,成功分離雷米普利試樣及雷米普利和喹那普利的混合試樣。所得色譜圖如圖3a和3b所示,混合試樣中各物質的出峰順序依次為:雷米普利、喹那普利,且雷米普利和喹那普利的分離度為2.00。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為2120。
對比實施例4
整體柱製備:
不加任何模板分子,將0.14ml功能單體甲基丙烯酸(MAA)溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比50:50)充分衝洗至基線平穩後,即製備得到非印跡整體柱。
分離效果測試:
以對比實施例4製備非印跡整體柱為固定相,在反相色譜模式下對雷米普利(1.5mg/ml)、苯那普利(3.0mg/ml)、喹那普利(3.0mg/ml)和卡託普利(0.5mg/ml)混合試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1.0ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,分離四種普利類藥物混合試樣。所得色譜圖如圖4所示,出峰順序依次為:卡託普利、雷米普利、苯那普利、喹那普利,卡託普利與雷米普利、雷米普利與苯那普利以及苯那普利與喹那普利之間的分離度分別為:2.68、1.04、0.86,與實施例1、2、3相比較,分離度明顯較差。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1693。
實施例5
整體柱製備:
將166.6mg模板分子雷米普利、0.14ml功能單體甲基丙烯酸(MAA)和0.05ml苯乙烯溶解在0.53ml N,N-二甲基甲醯胺和2.41ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣10min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比40:60)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例5製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.2)及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖5所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為2560塊/米。
實施例6
整體柱製備:
將33.3mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.53ml N,N-二甲基甲醯胺和3.18ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和40mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣30min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比90:10)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例6製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM pH 2.2磷酸二氫鉀(PBS)緩衝溶液及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖6所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1666塊/米。
實施例7
整體柱製備:
將100mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.53ml N,N-二甲基甲醯胺和2.41ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入1.25ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和30mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣10min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比10:90)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例7製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)及乙腈混合溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖7所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為2633塊/米。
實施例8
整體柱製備:
將167mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.53ml N,N-二甲基甲醯胺和2.41ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.55ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和10mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理20min,使其充分溶解並混合均勻,再通氮氣15min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合36h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比20:80)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例8製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM pH 2.5磷酸二氫鉀(PBS)緩衝溶液及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖8所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1280塊/米。
實施例9
整體柱製備:
將100mg模板分子雷米普利、0.14ml功能單體甲基丙烯酸(MAA)和0.07ml苯乙烯溶解在0.53ml N,N-二甲基甲醯胺和2.41ml十二醇中,室溫下超聲處理20min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理45min,使其充分溶解並混合均勻,再通氮氣15min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合36h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比70:30)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例9製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)和乙腈混合溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖9所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1633塊/米。
實施例10
整體柱製備:
將100mg模板分子雷米普利、0.14ml功能單體甲基丙烯酸(MAA)和0.14ml苯乙烯溶解在0.53ml N,N-二甲基甲醯胺和2.41ml十二醇中,室溫下超聲處理20min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣15min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合36h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比40:60)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例10製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)和乙腈混合溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖10所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為2320塊/米。
實施例11
整體柱製備:
將100mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和30mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比90:10)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例11製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣及雷米普利(1.0mg/ml)、苯那普利(1.0mg/ml)、喹那普利(1.0mg/ml)、卡託普利(1.0mg/ml)混合試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)及乙腈溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為1.0ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,成功分離雷米普利試樣及四種普利類藥物混合試樣。所得色譜圖如圖11a和11b所示,混合試樣中各物質的出峰順序依次為:卡託普利、雷米普利、苯那普利、喹那普利,且卡託普利與雷米普利、雷米普利與苯那普利以及苯那普利與喹那普利之間的分離度分別為3.26、1.46、1.15,具有較好的分離效果。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為3253塊/米。
實施例12
整體柱製備:
將33mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和30mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比30:70)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例12製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣及雷米普利(1.0mg/ml)、苯那普利(1.0mg/ml)、喹那普利(1.0mg/ml)、卡託普利(1.0mg/ml)的混合試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)和乙腈混合溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為0.8ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖12a和12b所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1027塊/米。
實施例13
整體柱製備:
將133mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸(MAA)溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和30mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比30:70)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例13製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣及雷米普利(1.0mg/ml)、苯那普利(1.0mg/ml)、喹那普利(1.0mg/ml)、卡託普利(1.0mg/ml)的混合試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)和乙腈混合溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為0.8ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖13a和13b所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1473塊/米。
實施例14
整體柱製備:
將100mg模板分子雷米普利和0.14ml功能單體甲基丙烯酸丁酯溶解在0.53ml N,N-二甲基甲醯胺和2.41ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和10mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於65℃的恆溫箱內進行熱引發聚合12h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比70:30)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例14製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)和乙腈混合溶液為流動相,乙腈:PBS=20:80(體積比),流動相流速為0.8ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖14所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1307塊/米。
實施例15
整體柱製備:
將100mg模板分子雷米普利和185mg(相當於0.165ml)功能單體丙烯醯胺溶解在0.44ml甲苯和2.00ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入0.77ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和40mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mm i.d.×150mm)中,兩端密封,於45℃的恆溫箱內進行熱引發聚合30h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比70:30)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例15製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)和甲醇混合溶液為流動相,甲醇:PBS=20:80(體積比),流動相流速為0.8ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖15所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1073塊/米。
實施例16
整體柱製備:
將100mg模板分子雷米普利和0.53ml功能單體甲基丙烯酸縮水甘油酯(GMA)溶解在1.10ml甲苯和1.73ml十二醇中,室溫下超聲處理30min,溶液充分混合後,加入1.13ml交聯劑乙二醇二甲基丙烯酸酯(EDMA)和20mg引發劑偶氮二異丁腈(AIBN),繼續室溫下超聲處理30min,使其充分溶解並混合均勻,再通氮氣5min,除去溶液中的溶解氧。隨後,將此溶液加入到一根不鏽鋼管(4.6mmi.d.×150mm)中,兩端密封,於55℃的恆溫箱內進行熱引發聚合18h。反應完成後,將其連接到高效液相色譜儀上,先用0.1ml/min甲醇衝洗0.5h,然後再分別用1ml/min的甲醇、甲醇醋酸溶液(體積比8:1)和水分別衝洗1h,洗去模板分子、致孔劑和未反應的溶劑,最後再用甲醇水溶液(體積比55:45)充分衝洗至基線平穩後,即製備得到雷米普利分子印跡整體柱。
分離效果測試:
以實施例16製備的雷米普利分子印跡整體柱為固定相,在反相色譜模式下,對雷米普利(1.0mg/ml)試樣進行分離。以20.0mM磷酸二氫鉀(PBS)緩衝溶液(pH 2.5)和乙腈混合溶液為流動相,乙腈:PBS=10:90(體積比),流動相流速為1.0ml/min,進樣量5μL,柱溫35℃,檢測波長為254nm,所得色譜圖如圖16所示。
該整體柱在甲醇:水=85:15的流動相條件下,用萘測得該雷米普利分子印跡整體柱的理論塔板數為1073塊/米。