一種α‑酮基丁酸的發酵生產工藝的製作方法
2023-05-03 05:12:46 3
本發明涉及化合物生物技術生產領域,尤其是一種α-酮基丁酸的發酵生產工藝。
背景技術:
α-酮基丁酸的生產方法包括化學合成法和生物合成法。化學法是由草酸二乙酯和丙酸乙酯混合水解得到α-酮基丁酸。生物法合成α-酮基丁酸主要為前體物轉化法和直接發酵法。Nakahara等(1994)以1,2-丁二醇為碳源和底物,採用馬紅球菌(Rhodococcus equi)IF03730將其轉化為α-酮基丁酸,培養32h轉化率為68.2%。隨後研究發現惡臭假單胞菌(Pseudomonas sputita)和施氏假單胞菌(Pseudomonas stutzeri)SDM分別能將巴豆酸和DL-2-羥基丁酸轉化為α-酮基丁酸,其產量為4.8g/L。馬翠卿等(ZL201110376233.8)報導了以L-蘇氨酸為底物利用含L-蘇氨酸脫水酶的假單胞菌、棒桿菌屬或芽孢桿菌屬為生物催化劑製備α-酮基丁酸的方法,轉化液中α-酮基丁酸的濃度為25.6g/L,轉化率為99.6%。陳寧等(201410132996.1)構建了過表達的蘇氨酸脫水酶編碼基因ilvA、敲除乙醯羥基酸合成酶Ⅰ大亞基編碼基因ilvB、乙醯羥基酸合成酶Ⅲ大亞基編碼基因ilvI以及蘇氨酸操縱子前導肽編碼基因thrL的基因工程大腸桿菌,該菌株以葡萄糖為底物發酵20-24h後,α-酮基丁酸最高產量達到15.7g/L。該方法通過低拷貝質粒pwsk29直接過表達蘇氨酸脫水酶編碼基因ilvA,且出發菌株為大腸桿菌E.col iMG1655標準株。
目前工業生產α-酮基丁酸主要採用化學合成法,但該方法存在反應條件複雜、能耗大、汙染重等問題。而生物法的前體物轉化法存在著底物生產成本高,微生物製劑難於培養等問題;生物法中利用重組大腸桿菌直接發酵生產α-酮基丁酸最大的問題在於產物毒性高,如圖1所示,α-酮基丁酸濃度達到8g/L以上時,α-酮基丁酸對大腸桿菌菌體生長和產物合成存在嚴重的抑制作用。同時通過實驗發現,敲除乙醯羥基酸合成酶Ⅰ基因ilvBN會影響纈氨酸的合成,菌體生物量大幅度下降,且對提升α-酮基丁酸的積累量無明顯作用。
技術實現要素:
本發明所要解決的技術問題在於提供一種α-酮基丁酸的發酵生產工藝,可高效製備α-酮基丁酸。
為解決上述技術問題,本發明的技術方案是:
一種α-酮基丁酸的發酵生產工藝,對大腸桿菌E.col iTHRD(保藏號CGMCC No.11074)蘇氨酸脫水酶編碼基因ilvA進行定點突變、解除L-異亮氨酸對ilvA的反饋抑制的基礎上過表達ilvA;敲除了乙醯羥基酸合成酶III的大亞基編碼基因ilvI;利用溫度控制誘導的方式,採用發酵法生產α-酮丁酸,其中,最佳誘導時機和最合適的升溫誘導模式為:35-37℃培養菌體,生長到OD600為10-15時,1h內升溫至40-42℃(快速升溫)。
優選的,上述α-酮基丁酸的發酵生產工藝,利用溫控誘導方式製備α-酮基丁酸,具體步驟如下:
Ⅰ、搖瓶發酵積累α-酮基丁酸
活化斜面培養:將產生α-酮基丁酸的基因工程菌劃線接於1支新鮮AmpR抗性斜面,37℃培養15-17h;
搖瓶種子培養:從新鮮活化斜面上挑取1環菌體接於30mL的種子培養基中,200r/min,35-37℃恆溫振蕩培養,種子培養期間根據指示劑苯酚紅顏色變化,用25%(W/V)氨水調節pH≈6.7-7.0;
搖瓶發酵培養:保證溶氧充足情況下,500mL擋板三角瓶裝液量30mL,9層紗布封口;待種子液培養至OD600值約為8-10時,種齡為10-12h,按8-10%接種量接入發酵培養基中,200r/min,32-35℃恆溫振蕩培養;待發酵4-6h,菌體OD600值約為10-15時,將發酵溫度從32-35℃直接提高到40-42℃開始升溫誘導,直至24h發酵結束,發酵期間補加25%(W/V)的氨水維持發酵液pH≈6.7-7.0;
上述步驟(3)所得α-酮基丁酸的產量可達到9.4-12.5g/L;
或
Ⅱ、7.5L發酵罐發酵積累α-酮基丁酸
活化斜面培養:將產生α-酮基丁酸的基因工程菌劃線接於4支新鮮AmpR抗性斜面,37℃培養16h左右;
7.5L罐種子培養:吸取適量滅菌的去離子水於4支新鮮的活化斜面,刮下所有菌體後將菌懸液接入裝有2L種子培養基的5L發酵罐;種子培養溫度為35-37℃,通氣量變化範圍為2-5L/min,攪拌轉速變化範圍為100-500r/min,期間通過流加25%(W/V)的氨水控制pH≈6.7-7.0;
7.5L罐發酵培養:種子液培養至OD600=10-15時,種齡約為7h,將500mL左右種子液轉移到含有3.5L新鮮無菌發酵培養基的7.5L發酵罐中,總裝液量為4L,接種量為8-10%;32-35℃開始進行發酵培養;待發酵液OD600值為20-25時,在0.5-1.0h內將發酵溫度從32-35℃提高到40-42℃開始升溫誘導,直至28h發酵結束,發酵過程中通過自動流加25%(W/V)氨水控制pH≈6.7-7.0,溶氧控制在30-50%,通氣量變化範圍為2-8L/min,攪拌轉速變化範圍為200-900r/min;當發酵培養基中葡萄糖耗完後,將80%(W/V)葡萄糖溶液按1-1.5mL/min的脈衝速度流加至培養基中以維持發酵液中殘糖濃度控制在0-1g/L;
上述步驟(3)所得α-酮基丁酸的產量可達到16.5-22.8g/L。
上述α-酮基丁酸的製備方法,所得發酵液中α-酮基丁酸的檢測方法為:發酵液樣品經13000轉離心2min,上清液經0.22μm膜過濾後用UltiMate3000(Thermo Scientific)高效液相測試α-酮丁酸產量,檢測條件各參數為:色譜柱aminexR HPX-87H(300mm×7.8mm),流動相為5mmol/L H2SO4,流速0.5mL/min,柱溫30℃,檢測波長215nm,進樣量為20μL。
優選的,上述α-酮基丁酸的發酵生產工藝,所述產生α-酮基丁酸的基因工程菌是以蘇氨酸生產菌株E.coli THRD(保藏號CGMCC No.11074)為出發菌株,利用熱誘導型表達載體PBV220、PBV221或PBV222過表達定點突變的蘇氨酸脫水酶編碼基因ilvA並敲除乙醯羥基酸合成酶III的大亞基編碼基因ilvI。
優選的,上述α-酮基丁酸的發酵生產工藝,所述定點突變的蘇氨酸脫水酶編碼基因ilvA的核苷酸序列為序列表2所示序列。
所述編碼基因ilvA原始(未突變)核苷酸序列為序列表1所示序列;所述編碼基因ilvI的核苷酸序列為序列表3所示序列;
優選的,上述α-酮基丁酸的發酵生產工藝,所述產生α-酮基丁酸的基因工程菌是由下述方法構建得到的,具體步驟為:
一、基因ilvI的敲除
採用PCR技術以大腸桿菌E.coli THRD(保藏號CGMCC No.11074)基因組為模板,根據E.col iMG1655中ilvI(GeneID:948793)基因的5』和3』端500bp序列設計同源臂引物,擴增獲取ilvI基因的上下遊同源臂;
採用PCR技術以pKD3質粒為模板,設計引物,擴增氯黴素抗性基因片段;
以、獲得的擴增片段為模板通過重疊PCR獲得ilvI基因的敲除片段,所述敲除片段由乙醯羥基酸合成酶III大亞基編碼基因ilvI的上、下遊同源臂基因片段以及氯黴素抗性基因片段組成;
將上述基因敲除片段電轉入含pKD46質粒的大腸桿菌E.coliTHRD感受態細胞中獲得陽性轉化子,消除掉陽性菌中的氯黴素抗性基因後即可獲得ilvI基因敲除菌;
二、ilvA基因的定點突變和過表達
採用PCR技術以大腸桿菌E.coli THRD(保藏號CGMCC No.11074)基因組為模板,根據E.col iMG1655中ilvA(GeneID:948287)基因的5』端到突變處設計引物ilvA-1、ilvA-2,突變處到3』端設計引物ilvA-3、ilvA-4,擴增獲取ilvA基因突變處前後的兩段片段A1和A2;
通過引物ilvA-2和ilvA-3完全反向互補,擴增片段重疊後將鹼基序列GCTGCTGCGCTTCCT突變為GTCTGCTGCGCTTCGC,即以中獲得的兩條擴增片段為模板通過重疊PCR獲得突變後的ilvA基因;
將點突變後的ilvA基因及溫控表達載體PBV220(也同樣可用於PBV221-ilvA或PBV222-ilvA)分別用pstI和BamHI進行雙酶切後連接,連接產物轉化至上述ilvI基因敲除菌中,菌落PCR鑑定後獲得的陽性轉化子即為本發明所述的產生α-酮基丁酸的基因工程菌;
所述PBV221-ilvA,PBV222-ilvA和PBV220-ilvA構建方法相同。
本發明的有益效果是:
上述α-酮基丁酸的發酵生產工藝,利用熱誘導型表達載體過表達定點突變的L-蘇氨酸脫水酶基因ilvA並敲除乙醯羥基酸合成酶III的大亞基編碼基因ilvI,通過溫度變化人為調控表達,操作簡便、成本低,在發酵過程中通過溫度控制將菌體的快速生長期和α-酮基丁酸合成期分開,使這兩個階段互不影響,從而解決發酵過程中高濃度的α-酮基丁酸(≧8g/L)嚴重抑制菌體生長和產酸能力等問題,達到提高菌體生物量,增加α-酮基丁酸發酵水平的目的。使用特定的產生α-酮基丁酸的基因工程菌,通過溫控誘導的發酵方式,可以更加有效的解決發酵過程中α-酮基丁酸對菌體生長和產酸的抑制問題。
附圖說明
圖1為背景技術中α-酮基丁酸濃度梯度下基因工程菌乾重曲線圖譜。
圖2為E.coli THRD△ilvIH的構建過程圖。
圖3-a為ilvI敲除重疊PCR圖譜:其中M為Marker;1為ilvI基因上遊498bp;3為氯黴素抗性基因片段1090bp;4為ilvI基因下遊497bp;
圖3-b為ilvI敲除鑑定PCR圖譜:其中M為Marker;1為E.coli THRD菌株1175bp;2為消除氯黴素抗性的ilvI敲除菌1545bp;3為成功消除氯黴素抗性的ilvI敲除菌500bp。
圖4為重組質粒PBV220-ilvA菌株構建過程圖。
圖5為重組質粒PBV220-ilvA酶切鑑定圖譜:其中M為Marker;1為BαmHI和PstI雙酶切PBV220空質粒3666bp;2為BαmHI和PstI雙酶切重組質粒PBV220-ilvA3666bp和1561bp。
圖6為α-酮基丁酸標準品(濃度10g/L)液相檢測峰圖。
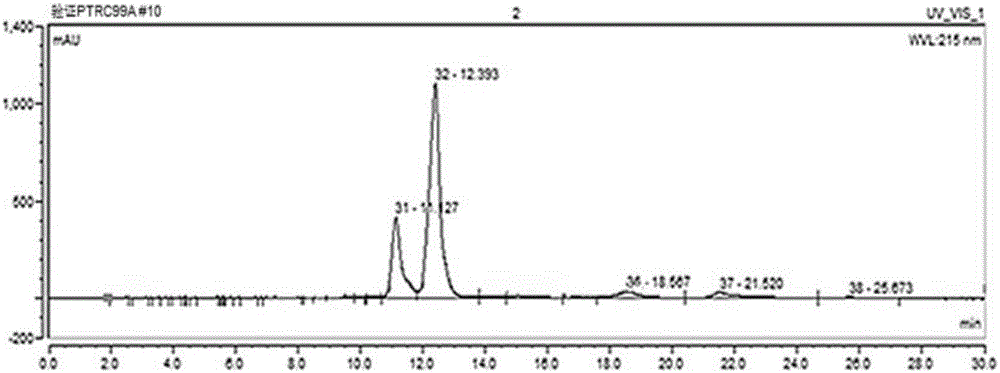
圖7為搖瓶發酵24h發酵液樣品液相檢測峰圖。
圖8為7.5L罐發酵28h發酵液樣品液相檢測峰圖。
圖9為7.5L發酵罐上α-酮基丁酸積累量的發酵過程曲線。
保藏信息
分類名詞:大腸埃希氏菌Escherichia coli
保藏單位名稱:中國微生物菌種保藏管理委員會普通微生物中心
保藏單位地址:北京市朝陽區北辰西路1號院3號
保藏日期:2015年7月14日
保藏號:CGMCC No.11074
具體實施方式
為了使本領域的技術人員更好的理解本發明的技術方案,下面結合具體實施方式對本發明所述技術方案作進一步的詳細說明。
實施例1:產生ɑ-酮基丁酸的基因工程菌株E.coli THRD△ilvIH+PBV220-ilvA的構建
1.乙醯羥基酸合成酶III大亞基編碼基因ilvI的敲除
(1)根據NCBI資料庫中E.col iMG1655中ilvI基因5』和3』端500bp序列設計上下遊同源臂擴增引物ilvI-1、ilvI-2和ilvI-5及ilvI-6,並以E.coliTHRD菌株(保藏號CGMCC No.11074)基因組DNA為模板擴增同源臂片段。PCR擴增條件為95℃5min1個循環,94℃30s、56℃30s、72℃1min25個循環,72℃10min1個循環,反應體系為50μL。
(2)根據質粒pKD3中氯黴素抗性基因序列設計出擴增引物CM-3、CM-4,並以質粒pKD3為模板擴增氯黴素抗性基因片段,PCR擴增條件為95℃5min1個循環,94℃30s、56℃30s、72℃1.5min25個循環,72℃10min1個循環,反應體系為50μL。將(1)和(2)中PCR產物經1.0%瓊脂糖凝膠電泳後切膠回收,將回收的片段分別命名為I1、CM2和I3。
(3)按1:2:1的摩爾濃度比混合I1、CM2和I3作為模板,利用引物ilvI-1、ilvI-6進行三段重疊PCR,PCR條件為95℃5min1個循環,94℃30s、56℃30s、72℃2.5min25個循環,72℃10min1個循環,反應體系為50μL。將重疊PCR產物經1.0%瓊脂糖凝膠電泳後切膠回收,獲得ilvI敲除片段,命名為△ilvIH。
(4)將片段△ilvIH電轉化至含pKD46質粒的E.coli THRD菌株(保藏號CGMCC No.11074)感受態細胞中(電轉化電壓為1800V,時間為5.5ms)。迅速加入1mL SOC培養基於37℃、200rpm復甦1h後塗布於含氯黴素(25μg/mL)LB固體培養基平板上。倒置培養24h後採用ilvI敲除鑑定引物△IH-F和△IH-R通過菌落PCR鑑定陽性轉化子,三段重疊片段成功整合到E.coli THRD基因組上的菌落其擴增片段約1454bp。將溫敏質粒pCP20轉化至上述陽性轉化子以去除氯黴素抗性基因,經42℃過夜培養後,篩選能夠在無抗性平板上生長而在含氯黴素平板上不生長的單菌落並採用ilvI敲除鑑定△IH-F和△IH-R進行驗證,ilvI被成功敲除且丟失氯黴素抗性基因的菌株擴增片段約為500bp,獲得E.coli THRD△ilvIH菌株。
構建過程如圖2,圖3-a,圖3-b和圖4為PCR驗證結果,引物序列見表1。
表1ilvI基因敲除引物
2.ilvA的定點突變與過表達
(1)根據NCBI資料庫中E.col iMG1655中ilvA基因序列設計ilvA-1、ilvA-2和ilvA-3、ilvA-4兩對引物,以E.coliTHRD基因組DNA為模板分別擴增A1和A2兩片段,PCR擴增條件為95℃5min1個循環,94℃30s、56℃30s、72℃1.5min25個循環,72℃10min1個循環,反應體系為50μL,將回收的片段分別命名為A1和A2。
(2)按1:1的摩爾濃度比混合A1和A2作為模板,利用引物ilvA-1、ilvA-4進行重疊PCR,PCR條件為95℃5min1個循環,94℃30s、56℃30s、72℃2min25個循環,72℃10min1個循環,反應體系為50μL。將重疊PCR產物經1.0%瓊脂糖凝膠電泳後切膠回收,獲得點突變的ilvA基因片段。
(3)將點突變的ilvA基因片段和質粒PBV220分別用PstI和BαmHI進行雙酶切後連接,連接產物化轉入大腸桿菌DH5α感受態細胞,挑取能在氨苄青黴素平板(100μg/mL)上生長的轉化子並經鑑定引物PBV-jd1和PBV-jd2進行菌落PCR鑑定。提取陽性轉化子質粒進行測序,結果與實驗預期一致,表明重組質PBV220-ilvA粒構建成功。構建過程如圖4,雙酶切電泳圖譜如圖5。
(4)將重組質粒PBV220-ilvA電轉入實施例1獲得的ilvIH基因單敲除菌株,挑取能在氨苄青黴素平板(100μg/mL)上生長的轉化子並經鑑定引物PBV-jd1和PBV-jd2進行菌落PCR鑑定,獲得的陽性轉化子即為本發明所述基因工程菌,命名為E.coli THRD△ilvIH+PBV220-ilvA。實例2所用引物序列見表2。
表2ilvA基因過表達引物
實施例2:產生α-酮基丁酸的基因工程菌的搖瓶發酵
菌株:E.coli THRD△ilvIH+PBV220-ilvA抗性:AmpR
種子培養:菌種經AmpR斜面活化兩代後,無菌條件下用接種針刮1環溼菌體接於30mL含有100μg/mLAmpR的種子培養基中。37℃,200rpm培養8h,期間根據指示劑顏色變化用氨水調pH≈7.0。
發酵條件:種子液培養8h後,種子液OD600=8,用滅菌的5mL移液管無菌條件下吸取3mL種子液接於27mL含有100μg/mLAmpR的發酵培養基中,接種量為10%。35℃,200rpm開始進行發酵培養,當發酵液OD600=15時,直接提高溫度至42℃進行誘導表達,至發酵24h結束,期間根據指示劑顏色變化,用氨水調節pH≈7.0。
利用高效液相色譜測定發酵液中α-酮基丁酸的含量,具體檢測方法為:發酵液樣品經13000轉離心2min,上清液經0.22μm膜過濾後用UltiMate3000(Therm Scientific)高效液相測試α-酮丁酸產量,檢測條件各參數為:色譜柱AminexR HPX-87H(300mm×7.8mm),流動相為5mmol/L H2SO4,流速0.5mL/min,柱溫30℃,檢測波長215nm,進樣量為20μL。經檢測,如圖6和圖7所示,α-酮基丁酸搖瓶產量為12.5g/L。
其中,種子培養基(g/L):葡萄糖25,酵母粉10,蛋白腖6,KH2PO41.2,MgSO4·7H2O 0.5,FeSO4·7H2O 10mg/L,MnSO4·H2O 10mg/L,VB11.3mg/L,VH 0.3mg/L,pH 6.7-7.0,115℃滅菌15min。
發酵培養基(g/L):葡萄糖30,酵母粉2,蛋白腖4,檸檬酸鈉1,KH2PO42,MgSO4·7H2O 0.7,FeSO4·7H2O 100mg/L,MnSO4·H2O 100mg/L,VB11.3mg/L,VH 0.3mg/L,pH=6.7-7.0,消泡劑若干滴,121℃滅菌20min。
實施例3:生產α-酮基丁酸基因工程菌的7.5L發酵罐發酵
菌株:E.coli THRD△ilvIH+PBV220-ilvA抗性:AmpR
種子培養:菌種經AmpR斜面活化兩代後,無菌條件下用接種針將4支斜面上的溼菌體全部接種到裝有2L種子培養基的5L種子罐中,流加25%(W/V)的氨水調節種子液pH=7.0,通過提高轉速和通風量使溶氧維持在30-50%左右,37℃恆溫培養。
發酵條件:待種子液OD600=15時,將500mL種子液轉移到含有無菌發酵培養基的7.5L發酵罐中,總裝液量為4L,接種量為12%。35℃培養,期間流加25%(W/V)的氨水調節發酵液pH≈6.7-7.0,提高轉速和通風量使溶氧維持在30-50%左右。待發酵液OD600=25時,採用35℃→37℃→39℃→40℃(注意0.5h內完成)的升溫模式進行誘導表達,之後40℃恆溫發酵培養至28h發酵結束。
其中種子培養基和發酵培養基配方同實施例2中所述。
利用高效液相色譜測定發酵液中α-酮基丁酸的含量,具體檢測方法同實施例2中所述。經檢測,如圖8和圖9所示,α-酮基丁酸的上罐產量為22.8g/L。
上述參照具體實施方式對該一種α-酮基丁酸的發酵生產工藝進行的詳細描述,是說明性的而不是限定性的,可按照所限定範圍列舉出若干個實施例,因此在不脫離本發明總體構思下的變化和修改,應屬本發明的保護範圍之內。