作為抗結核劑的新化合物的製作方法
2023-07-15 21:54:26 2
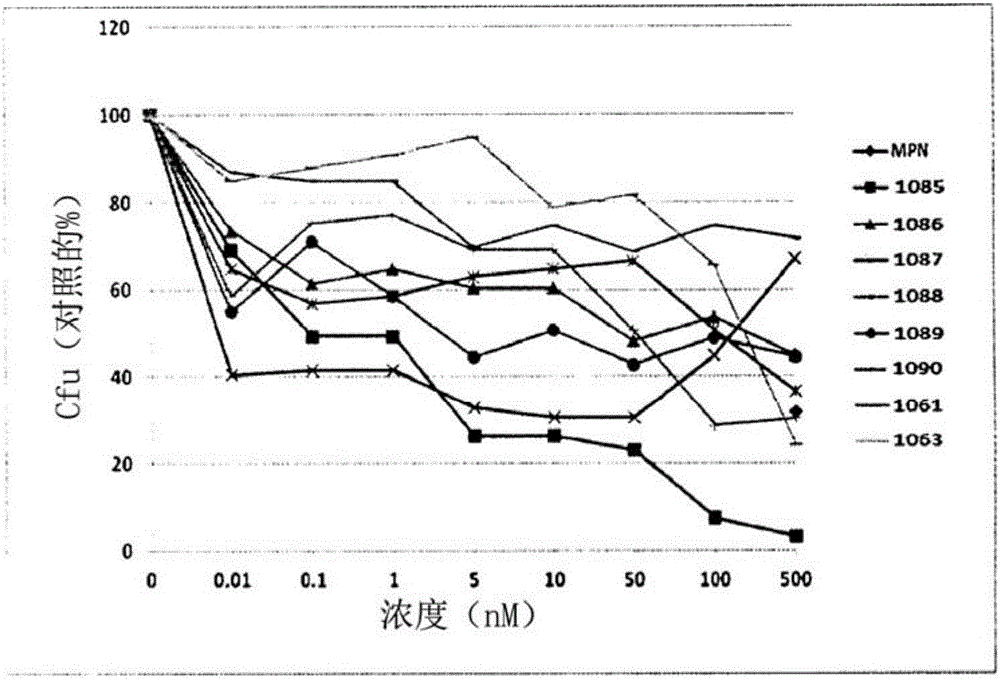
本發明涉及用作抗結核劑(抗結核病藥劑,anti-tubercularagent)的新化合物。更具體地,本發明涉及取代的衍生物、其製備方法、含有這些化合物的藥物組合物以及這些化合物在治療結核病中的用途。
背景技術:
:結核病(肺結核,tuberculosis)每年導致近兩百萬人的死亡。近些年已經見證了針對結核病(TB)藥物研究和開發的努力重新出現。對抗結核治療年表(大事記,chronology)的分析表明了對結核病研究恢復的興趣。自二十世紀六七十年代以來,即使隨著耐藥性菌株的發展,存在對所述藥物的抗性,同一代的抗生素或它們的衍生物也被用於抗結核分枝桿菌;該耐藥性菌株為諸如對至少異煙肼(INH)和利福平(RMP)(兩種最強有力的一線治療抗TB藥物)具有抗性的多藥物耐性TB(MDR-TB),對異煙肼和利福平具有抗性的廣譜耐藥性TB(XDR-TB),加上任何氟喹諾酮和三種可注射的二線藥物(諸如阿米卡星、卡那黴素或捲曲黴素)中的至少一種,以及對測試的所有一線和二線藥物(諸如異煙肼、利福平、鏈黴素、乙胺丁醇、吡嗪醯胺、乙硫異煙胺、對氨基水楊酸、環絲氨酸、氧氟沙星、阿米卡星、環丙沙星、捲曲黴素、卡那黴素)具有抗性的完全耐藥性TB(TDR-TB)。印度是世界上結核病負擔最重的國家之一,其中MDR結核病每年佔近五十萬例,在過去10年中增長了大於6%。據說結核分枝桿菌(M.tuberculosis)通過其基因組的誘導或自發的突變而產生對藥物的抗性。還假設在某些藥物的情況下,細菌的細胞壁不允許足夠的通透性,從而導致不足的/少量的藥物進入細菌。這樣的量不足以殺死細菌,但是少量的存在會誘導該細菌變得對其產生抗性。還假設結核分枝桿菌通過未知的機制修飾其酶而獲得抗性。通過口服途徑給藥的傳統抗結核藥物通過抑制黴菌酸(分枝菌酸,mycolicacid)的合成和/或通過抑制分枝桿菌阿拉伯糖基轉移酶(分枝桿菌細胞壁的基本組分(主要成分))起作用。很少的全身性抗結核藥物也通過穿過脂質雙層並結合到核糖體亞基中的一個而起作用,從而抑制蛋白質合成。基於這些藥物的作用機制,這些藥物具有誘發突變的高概率和/或具有滲透性的問題,從而增加耐藥菌株的機會。諸如GPR109A的G蛋白偶聯受體在結核病中也起到重要作用。GPR109A受體位於細胞表面並抑制腺苷酸環化酶,隨之抑制PKA信號轉導,造成甘油三酯轉化減少,這進一步造成脂質體在細胞內的積累。細胞內脂質體濃度的增加有利於結核分枝桿菌的生長並防止呼吸爆發(respiratoryburst)。GPR109A抑制劑通過為結核病形成不利的環境而防止脂質體的形成,並從而誘導呼吸爆發。被結核分枝桿菌感染的宿主巨噬細胞獲得泡沫表現型,其特徵在於通過調節中性脂質的脂解(脂解作用,lipolysis)而由病原體誘導的脂質體的細胞內積累。這種調節異常通過調節cAMP依賴性信號通路來影響脂解。目前抗結核的治療具有更大的副作用風險並持續18-24個月,但是結核分枝桿菌內的靶過程或酶遭受產生顯示耐藥性的更新變體的風險。由於結核分枝桿菌通過調節一系列宿主細胞過程而在人類巨噬細胞內存活,因此,已經為其存活而被結核分枝桿菌指定(共同選擇)的宿主內的藥理性靶點應該是用於治療的突破性方法。另外,這種方法應該不受對感染菌株是否是藥物敏感的還是耐藥的影響,並且阻止抗性的發展。發明目的本發明的一個目的是提供可用作抗結核劑的化合物。技術實現要素:本發明提供了以下式1的新化合物、以及其鹽、水合物和立體異構體:其中,X選自O、NH或N(烷基);R1和R2獨立地選自氫、氘、羥基、C1-10直鏈或支鏈烷基、3-7元環烷基、C1-6烷氧基、芳基、氨基、NH(烷基)、N(烷基)2、OCOR5、含有1-3個選自包括O、N或S的組的雜原子的雜芳基;或者R1和R2可以被組合以形成含有1-3個選自包括O、N或S的組的雜原子的芳基或雜芳基環;R3選自氫、羥基、C1-6直鏈或支鏈烷基、3-7元環烷基、C1-6烷氧基、芳基、選自以下結構式的芳香族或非芳香族雜環或稠合雜環(稠雜環):X可以與R3一起形成包含1-3個選自包含N、O或S的組的雜原子的5-7元雜環。所述雜環進一步被一個或多個低級烷基、滷素、氨基、NH(烷基)、N(烷基)2、NH-芳烷基取代;R4選自氫、低級直鏈或支鏈烷基、滷素、氘、C1-6烷氧基、氨基、NH(烷基)、N(烷基)2、-COOR8、CONR8R9;R5是氫、羥基、C1-6烷基、C1-6烷氧基、氨基、NH(烷基)、N(烷基)2;R6和R7獨立地選自包括氫、C1-10烷基、COR8、-CH2OCOR8、-CH2OCONHR8R9、-COOR8、-CONR8R9、-SO2R8、芳基、芳烷基的組;R8和R9獨立地選自包括氫、或C1-6直鏈或支鏈烷基的組;n為1、2或3。附圖說明圖1描述了本發明所述化合物在MYC-431感染的THP-1巨噬細胞中的作用。圖2描述了本發明的化合物對脂質體的作用。圖3描述了本發明的化合物1085在細胞M.tb中的效力。圖4表示了化合物1085對THP-1巨噬細胞中腺苷酸環化酶(c-AMP)的作用。圖5a和圖5b表示了結核分枝桿菌與降解性囊泡(degradativevesicle)的共定位:在化合物1085存在下的酸化溶酶體和自噬體。圖6表示了化合物1085對細胞脂質體的作用。圖7表示了化合物1085對多種分枝桿菌菌株的作用。圖8表示了化合物1085單獨對分枝桿菌生長和與其他藥劑組合對分枝桿菌生長的作用。圖9a和圖9b表示了通過腹膜內途徑給藥的化合物1085的藥代動力學參數。圖10表示了化合物1085與抗結核治療(ATT)的聯合治療。具體實施方式A.本發明的化合物:因此,本發明提供了作為抗結核病藥劑的式1的新化合物。本發明涉及式(1)的新化合物、及其鹽、水合物和立體異構體:其中,X選自O或NH;R1和R2獨立地選自氫、氘、羥基、C1-10直鏈或支鏈烷基、3-7元環烷基、C1-6烷氧基、芳基、氨基、NH(烷基)、N(烷基)2、OCOR5、含有1-3個選自包括O、N或S的組的雜原子的雜芳基;或者R1和R2可以被組合以形成含有1-3個選自包括O、N或S的組的雜原子的芳環或雜芳環;R3選自氫、羥基、C1-6直鏈或支鏈烷基、3-7元環烷基、C1-6烷氧基、芳基、選自以下結構式的芳香族或非芳香族雜環或稠合雜環:X可以與R3一起形成包含1-3個選自包含N、O或S的組的雜原子的5-7元雜環。所述雜環進一步被一個或多個低級烷基、滷素、氨基、NH(烷基)、N(烷基)2、NH-芳烷基取代;R4選自氫、低級直鏈或支鏈烷基、滷素、氘、C1-6烷氧基、氨基、NH(烷基)、N(烷基)2、-COOR8、CONR8R9;R5是氫、羥基、C1-6烷基、C1-6烷氧基、氨基、NH(烷基)、N(烷基)2;R6和R7獨立地選自包括氫、C1-10烷基、-COR8、-CH2OCOR8、-CH2OCONHR8R9、-COOR8、-CONR8R9、-SO2R8、芳基、芳烷基的組;R8和R9獨立地選自包括氫、或C1-6直鏈或支鏈烷基的組;n為1、2或3。此外,本發明涉及式(1)的新化合物、以及其鹽、水合物和立體異構體:其中X是NH;R1和R2選自氫、羥基、C1-10烷基、C1-6烷氧基;R3選自選自以下的取代的芳香族或非芳香族雜環或稠合雜環:R4選自氫、氘、滷素、C1-6直鏈或支鏈烷基、C1-6烷氧基、氨基、NH(烷基)、N(烷基)2;R6和R7獨立地選自氫、C1-10烷基、-COR8、-CH2OCOR8、-CH2OCONHR8R9、-COOR8、-CONR8R9、-SO2R8、苯基、苄基;R8和R9獨立地選自氫或C1-6直鏈或支鏈烷基;n為1、2或3。術語「烷基」是指直鏈或支鏈飽和一價烴,其中亞烷基可以可選地如本文所述的被取代。除非另有說明,否則術語「烷基」還包括直鏈和支鏈烷基。在某些實施方式中,所述烷基是具有特定數目的碳原子的直鏈飽和一價烴,或特定數目碳原子的支鏈飽和一價烴。如本文所使用的,直鏈C1-C6和支鏈C3-C6烷基也被稱為「低級烷基」。烷基的實例包括但不限於甲基、乙基、丙基(包括所有的同分異構形式)、正丙基、異丙基、丁基(包括所有的同分異構形式)、正丁基、異丁基、仲丁基、叔丁基、戊基(包括所有的同分異構形式)和己基(包括所有的同分異構形式)。例如,C1-C6烷基是指1至6個碳原子的直鏈飽和一價烴或3至6個碳原子的支鏈飽和一價烴。術語「芳基」是指含有至少一個芳香族烴環的單環芳香族基團和/或多環一價芳香族基團。在某些實施方式中,所述芳基具有6至20個環原子(C6-C20)、6至15個環原子(C6-C15)或6至10個環原子(C6-C10)。芳基的實例包括但不限於苯基、萘基、芴基、薁基、蒽基、菲基、芘基、聯苯基和三聯苯基。芳基也指雙環或三環碳環,其中所述環中的一個環是芳香族的,並且所述環中的其他環可以是飽和的、部分不飽和的或芳香族的,例如二氫萘基、茚基、茚滿基或四氫萘基(四氫萘基)。在某些實施方式中,芳基可以可選地如本文所述的被取代。術語「烷氧基」是指基團R'O--,其中R'是烷基。術語「低級烷氧基」是指具有1至3個碳原子的烷氧基;實例包括甲氧基、乙氧基、異丙氧基等。如本文所用的術語「環烷基」是指含有3至12個環原子或所指示的原子數的飽和或部分不飽和的單環、稠合雙環或橋連多環系統(橋接多環集合)。環烷基可以包括任何數目的碳,例如C3-6、C4-6、C5-6、C3-8、C4-8、C5-8和C6-8。飽和的單環環烷基環包括例如環丙基、環丁基、環戊基、環己基和環辛基。飽和雙環和多環環烷基環包括,例如降冰片烷、[2.2.2]雙環辛烷、十氫萘和金剛烷。環烷基也可以是部分不飽和的,在所述環中具有一個或多個雙鍵。部分不飽和的代表性環烷基包括,但不限於環丁烯、環戊烯、環己烯、環己二烯(1,3-異構體和1,4-異構體)、環庚烯、環庚二烯、環辛烯、環辛二烯(1,3-異構體、1,4-異構體和1,5-異構體)、降冰片烯和降冰片二烯。除非另有說明,否則環烷基是未取代的。「取代的環烷基」基團可以被一種或多種選自滷素、羥基、氨基、烷氨基、硝基、氰基和烷氧基的部分取代。術語「芳烷基」或「芳基-烷基」是指被芳基取代的一價烷基。在某些實施方式中,所述烷基和芳基部分如本文所述的可選地被取代。術語「雜芳基」是指含有至少一個芳環的單環芳基和/或多環芳基,其中至少一個芳環含有一個或多個獨立地選自O、S和N的雜原子。雜芳基的每個環可以含有一個或兩個O原子、一個或兩個S原子和/或1-4個N原子,條件是每個環中雜原子的總數為4或更少,並且每個環含有至少一個碳原子。在某些實施方式中,雜芳基具有5至20個、5至15個或5至10個環原子。單環雜芳基的實例包括,但不限於呋喃基、咪唑基、異噻唑基、異噁唑基、噁二唑基、噁二唑基、噁唑基、吡嗪基、吡唑基、噠嗪基、吡啶基、嘧啶基、吡咯基、噻二唑基、噻唑基、噻吩基、四唑基、三嗪基和三唑基。雙環雜芳基的實例包括,但不限於苯並呋喃基、苯並咪唑基、苯並異噁唑基、苯並吡喃基、苯並噻二唑基、苯並噻唑基、苯並噻吩基、苯並苯硫基(benzothiophenyl)、苯並三唑基、苯並噁唑基、呋喃並吡啶基、咪唑並吡啶基、咪唑並噻唑基、吲嗪基、吲哚基、吲唑基、異苯並呋喃基、異苯並噻吩基、異吲哚基(異氮(雜)茚基,isoindolyl)異喹啉基、異噻唑基、萘啶基、噁唑並吡啶基、酞嗪基、蝶啶基、嘌呤基、吡啶並吡啶基、吡咯並吡啶基、喹啉基、喹喔啉基、喹唑啉基、噻二唑並嘧啶基和噻吩並吡啶基。三環雜芳基的實例包括,但不限於吖啶基、苯並吲哚基、咔唑基、二苯並呋喃基、呸啶基、菲咯啉基、菲啶基、吩砷嗪基(phenarsazinyl)、吩嗪基、吩噻嗪基、吩噁嗪基和呫噸基(xanthenyl)。在某些實施方式中,雜芳基也可以可選地如本文所述的被取代。術語雜芳烷基是指如上所定義的芳烷基,其中一個或多個(優選地1個、2個、3個或4個)碳原子已經被氧、氮、矽、硒、磷、硼或硫原子(優選地氧、硫或氮)取代,也就是說根據上述定義的基團包含芳基或雜芳基和烷基、烯基、炔基和/或雜烷基和/或環烷基和/或雜環烷基的基團。雜芳烷基優選地含有一個或兩個具有5或6至10個碳原子和一個或兩個具有1或2至6個碳原子的烷基、烯基和/或炔基和/或具有5或6個環碳原子的環烷基的芳環體系(1或2個環),其中這些碳原子中的1個、2個、3個或4個已經被氧、硫或氮原子取代。實例是芳基-雜烷基、芳基-雜環烷基、芳基-雜環烯基、芳基烷基-雜環烷基、芳基烯基-雜環烷基、芳基炔基-雜環烷基、芳基烷基-雜環烯基、雜芳基烷基、雜芳基烯基、雜芳基炔基、雜芳基-雜烷基、雜芳基環-烷基、雜芳基環烯基、雜芳基-雜環烷基、雜芳基-雜環烯基、雜芳基烷基環烷基、雜芳基烷基-雜環烯基、雜芳基-雜烷基-環烷基、雜芳基-雜烷基環烯基和雜-芳基-雜烷基-雜環烷基,所述環基團是飽和的或單-、雙-或三-不飽和的。具體實例是四氫異喹啉基,苯甲醯基,2-或3-乙基吲哚基,4-甲基吡啶基,2-、3-或4-甲氧基苯基,4-乙氧基苯基和2-、3-或4-羧基苯基烷基。術語「雜環基」或「雜環的」是指含有至少一個非芳環的單環非芳香環系統和/或多環環系統,其中一個或多個非芳香族環原子是獨立地選自O、S或N的雜原子;並且剩餘的環原子是碳原子。在某些實施方式中,所述雜環基或雜環基團具有3至20個、3至15個、3至10個、3至8個、4至7個或5至6個環原子。在某些實施方式中,所述雜環基是單環、雙環、三環或四環環系統,其可以包括稠合或橋環系統,並且其中所述氮原子或硫原子可以可選地被氧化,所述氮原子可以可選地被季銨化,並且一些環可以是部分或完全飽和的,或是芳香族的。所述雜環基可以在任何雜原子或碳原子處連接至主結構,其導致穩定化合物的生成。這樣的雜環化合物的實例包括但不限於,氮雜基、苯並二噁烷基、苯並間二氧雜環戊烯基、苯並呋喃酮基、苯並吡喃酮基、苯並吡喃基、苯並四氫呋喃基、苯並四氫噻吩基、苯並噻喃基、苯並噁嗪基、對咔啉基、苯並二氫吡喃基、苯並二氫吡喃酮基、鄰二氮萘基、香豆素基、十氫異喹啉基、二氫苯並異噻嗪基、二氫苯並異噁嗪、二氫呋喃基、二氫異吲哚基、二氫吡喃基、二氫吡唑基、二氫吡嗪基、二氫吡啶基、二氫嘧啶基、二氫吡咯基、二氧戊環基、1,4-二噻烷基、呋喃酮基、咪唑烷基、咪唑啉基、二氫吲哚基、異苯並四氫呋喃基、異苯並四氫噻吩基、異苯並二氫吡喃基、異香豆素基、異吲哚啉基、異噻唑烷基、異噁唑烷基、嗎啉基、八氫吲哚基、八氫異吲哚基、噁唑烷酮基、噁唑烷基、環氧乙烷基、哌嗪基、哌啶基、4-哌啶酮基、吡唑烷基、吡唑啉基、吡咯烷基、吡咯啉基、奎寧環基、四氫呋喃基、四氫異喹啉基、四氫吡喃基、四氫噻吩基、硫嗎啉基、噻唑烷基、四氫喹啉基和1,3,5-三噻烷基。在某些實施方式中,雜環也可如本文所述的可選地被取代。術語「滷素」、「滷化物」或「滷代(halo)」是指氟、氯、溴和碘。如本文所用的術語「雜原子」是指除碳或氫之外的任何元素的原子。優選的雜原子為氮、氧、硫、磷和硒。如本文所用的術語「芳香族的」是指具有芳香族特性的不飽和環狀部分。該術語旨在包括烴芳香族化合物和雜芳香族化合物。術語「烴芳香環」或「烴芳香族化合物」是指這樣的芳香環或化合物,其中芳香族部分僅具有碳原子和氫原子。術語「雜芳環」或「雜芳香族化合物」是指這樣的芳香環或芳香族化合物,其中在至少一個芳香族部分中,環基團內的一個或多個碳原子已經被諸如氮、氧、硫等的另一個原子取代。如本文所用的術語「非芳香族的」是指環狀部分,其可以是不飽和的,但不具有芳香族特性。術語「取代的」是指分子上的氫自由基(hydrogenradical)已被另一原子自由基、官能團自由基或部分自由基取代;這些自由基(基團,radical)通常被稱為「取代基」。術語「可選取代的」意指基團(諸如烷基、亞烷基、烯基、亞烯基、炔基、亞炔基、烷氧基、烷氨基、二烷氨基、甲醯氨基、環烷基、亞環烷基、芳基、亞芳基、雜芳基、雜亞芳基、雜環基或雜亞環基)可被一個或多個取代基取代,所述取代基獨立地選自,例如(a)C1-C6烷基、C2-C6烯基、C2-C6炔基、C3-C7環烷基、C6-C14芳基、C7-C15芳烷基、雜芳基和雜環基,每種可選地被一個或多個取代基取代;和(b)滷素、氰基(-CN)、硝基(-NO2)、-C(O)R3、-C(O)OR3、-C(O)NRbRC、-C(NR3)NR)RC、-OR3、-OC(O)R3、-OC(O)OR3、-OC(O)NRbRC、-OC(=NR3)NR)RC、-OS(O)R3、-OS(O)2R3、-OS(O)NRbRC、-OS(O)2NRbRc、-NRbRc、-NR3C(O)Rd、-NR3C(O)ORd、-NR3C(O)NRbRC、-NR3C(=NRd)NRbRC、-NR3S(O)Rd、-NR3S(O)2Rd、-NR3S(O)NRbRC、-NR3S(O)NRbRc、-SR3、-S(O)R3、-S(O)2R3、-S(O)NRbRC和-S(O)2NRbRC,其中每個R3、Rb、Re和Rd獨立地是(i)氫;(ii)C1-C6烷基、C2-C6烯基、C2-C6炔基、C3-C7環烷基、C6-C14芳基、C7-C15芳烷基、雜芳基或雜環基,每種可選地用一個或多個取代基取代;或(iii)Rb和Re連同與它們相連接的N原子一起形成雜芳基或雜環基,可選地用一個或多個,在一個實施方式中,用一個、兩個、三個或四個取代基取代。除非另有說明,否則如本文所用的,可被取代的所有基團是「可選取代的」。在描述本發明的上下文中(特別是在所附權利要求的上下文中),使用術語「一個(一種(a))」和「一個(一種(an))」和「該」以及類似的指代應當被解釋為覆蓋單數和複數,除非在本文中另有說明或通過上下文明確禁用。如本文所採用的術語「一種或多種鹽」表示與無機酸和/或有機酸以及鹼形成的酸式鹽和/或鹼式鹽。兩性離子(內部的鹽或內鹽)包括在如本文使用的術語「一種或多種鹽」內(並且可以形成,例如,其中R取代基包含鹼性部分諸如氨基)。本文還包括季銨鹽,諸如烷基銨鹽。藥學上可接受的(即,無毒的、生理學上可接受的)鹽是優選的。術語「藥學上可接受的鹽」是指與合適的酸形成的酸加成鹽化合物,所述酸選自無機酸諸如鹽酸、氫溴酸;或有機酸諸如苯磺酸、馬來酸、草酸、富馬酸、琥珀酸、對甲苯磺酸和蘋果酸。如本文所用的術語「水合物」表示結晶分子化合物,其中水分子被結合到晶格中。一般來說,水合物因此表示分子化合物的結晶形式,其中被結合到晶格中的僅有的另外的分子是水分子。術語「立體異構體的」是指具有相同分子式和原子的連接性,但在三維空間中具有不同原子排列的至少兩種化合物。考慮到本公開內容,立體異構體可以是例如對映異構體、非對映異構體或內消旋化合物。如本文所用的術語「預防性的」是指預防和/或有助於預防感染的各種藥物、量或數量、方法、用途和效果等。如本文所用的術語「治療性的」是指預防、緩解(減輕)、治療、改善或治癒疾病或病症。術語「直接觀察的治療短程」(DOTS)是指由四種藥物(諸如利福平、異煙肼、乙胺丁醇和吡嗪醯胺)強化期兩個月組成的治療療程(方案),隨後是四個月的雙藥物(利福平和異煙肼)延續期。如本文所用的術語「多藥耐藥結核病」(MDR-TB)是指耐受至少異煙肼(INH)和利福平(RMP)(兩種最強大的一線治療抗結核藥物)的結核病。如本文所用的術語「廣泛耐藥性結核病」(XDR-TB)是指對六種二線抗結核藥物中的至少三種以及異煙肼和利福平,加上任何的氟喹諾酮和三種可注射的二線藥物(諸如阿米卡星、卡那黴素或捲曲黴素)中的至少一種具有耐性的結核病。如本文所用的術語「完全耐藥性結核病」(TDR-TB)是指對測試的所有一線和二線藥物(諸如異煙肼、利福平、鏈黴素、乙胺丁醇、吡嗪醯胺、乙硫異煙胺、對氨基水楊酸、環絲氨酸、氧氟沙星、阿米卡星(amikacin)、環丙沙星、捲曲黴素、卡那黴素)都有耐性的結核病。如本文所用的術語「H37Rv」是指已經測序的致病性結核分枝桿菌菌株。如本文所用的術語「JAL2287」是指結核分枝桿菌的MDR(多藥耐藥)菌株。如本文所用的術語「MYC431」是指結核分枝桿菌的XDR(廣泛耐藥)菌株。如本文所用的術語「GPR109A」是指G-蛋白偶聯受體GPR109A。本發明提供了由分子式(1)表示的化合物,其單獨或與DOTS(直接觀察的治療短程療法)治療組合抑制分枝桿菌集落生長。本發明的化合物可被舉例說明,但不限於如表1中提供的實例。表1:本發明的示例性化合物本發明還提供了如下分子式(1)的化合物:i.2-羥基-2,2-二苯基乙酸-1-甲基哌啶-3-基酯;ii.3-(2-羥基-2,2-二苯基乙醯氧基)-1-(((異丙基氨基甲醯基)氧基)甲基)-1-甲基哌啶-1-鎓iii.2-羥基-2,2-二苯基乙酸哌啶-3-基酯;iv.2-羥基-2,2-二苯基乙酸-1-苄基哌啶-3-基酯;v.2-羥基-2,2-二苯基乙酸-1-(甲磺醯基)哌啶-3-基酯;vi.2-羥基-2,2-二苯基乙酸-1-(二甲基氨基甲醯基)哌啶-3-基酯;vii.2-羥基-2,2-二苯基乙酸-1-苄基哌啶-4-基酯;viii.3-(2-羥基-2,2-二苯基乙醯氧基)-1-甲基奎寧環-1-鎓;ix.2-羥基-2,2-二苯基乙酸-1-苄基吡咯烷-3-基酯;x.2-羥基-2,2-二苯基乙酸-1-乙基哌啶-3-基酯;xi.2-羥基-2,2-二苯基乙酸-1-(異丙氨基甲醯基)哌啶-3-基酯;xii.2-羥基-2,2-二苯基乙酸-1-丙基哌啶-3-基酯;xiii.2-羥基-2,2-二苯基乙酸-1-甲基哌啶-4-基酯;xiv.2-羥基-2,2-二苯基乙酸-1-苄基氮雜環丁烷-3-基酯;xv.2-羥基-2,2-二苯基乙酸-1-乙醯基哌啶-3-基酯;xvi.2-羥基-2,2-二苯基-N-(哌啶-3-基)乙醯胺xvii.N-(1-苄基哌啶-4-基)-2-羥基-2,2-二苯基乙醯胺;xviii.2-羥基-2,2-二苯基乙酸-1-甲基吡咯烷-3-基酯;xix.2-羥基-2,2-二苯基乙酸奎寧環-3-基酯;xx.2-羥基-N-(1-甲基哌啶-3-基)-2,2-二苯基乙醯胺;xxi.N-(1-苄基哌啶-4-基)-2-羥基-2,2-二苯基乙醯胺;xxii.N-(1-苄基氮雜環丁烷(吖丁啶)-3-基)-2-羥基-2,2-二苯基乙醯胺;xxiii.2-羥基-2,2-二苯基乙酸氮雜環丁烷-3-基酯;xxiv.(S)-2-羥基-2,2-二苯基乙酸-1-苄基哌啶-3-基酯;xxv.(R)-2-羥基-2,2-二苯基乙酸-1-苄基哌啶-3-基酯;xxvi.(S)-2-羥基-2,2-二苯基乙酸-1-甲基哌啶-3-基酯;xxvii.(R)-2-羥基-2,2-二苯基乙酸-1-甲基哌啶-3-基酯;xxviii.(R)-N-(1-苄基哌啶-3-基)-2-羥基-2,2-二苯基乙醯胺;xxix.(R)-N-(1-苄基哌啶-3-基)-2-羥基-2,2-二苯基乙醯胺;xxx.2-羥基-2,2-二苯基-N-(哌啶-4-基)乙醯胺;xxxi.(S)-2-羥基-N-(1-甲基哌啶-3-基)-2,2-二苯基乙醯胺;xxxii.(R)-2-羥基-N-(1-甲基哌啶-3-基)-2,2-二苯基乙醯胺;xxxiii.2-羥基-2-苯基丙酸-1-苄基氮雜環丁烷-3-基酯;xxxiv.(R)-N-(1-苄基哌啶-3-基)-2,2-二苯基乙醯胺;xxxv.N-((S)-1-苄基哌啶-3-基)-2-羥基-2-苯基乙醯胺;xxxvi.N-((R)-1-苄基哌啶-3-基)-2-羥基-2-苯基丙醯胺;xxxvii.(R)-(R)-2-羥基-2-苯基乙酸-1-甲基哌啶-3-基酯;xxxviii.(R)-(S)-2-羥基-2-苯基乙酸-1-甲基哌啶-3-基酯;xxxix.(S)-(R)-2-羥基-2-苯基乙酸-1-甲基哌啶-3-基酯;xl.(S)-(S)-2-羥基-2-苯基乙酸-1-甲基哌啶-3-基酯;xli.(R)-2-羥基-2-苯基乙酸-1-甲基哌啶-4-基酯;xlii.(S)-2-羥基-N-(1-甲基哌啶-4-基)-2-苯基乙醯胺;xliii.(R)-2-羥基-N-(1-甲基哌啶-4-基)-2-苯基乙醯胺;xliv.(R)-2-羥基-N-((R)-1-甲基哌啶-3-基)-2-苯基乙醯胺;xlv.(R)-2-羥基-N-((S)-1-甲基哌啶-3-基)-2-苯基乙醯胺;xlvi.(S)-2-羥基-2-苯基-N-(吡啶-3-基)乙醯胺;xlvii.(S)-2-羥基-2-苯基乙酸-1-甲基哌啶-4-基酯;xlviii.(S)-2-羥基-2-苯基乙酸-1-苄基氮雜環丁烷-3-基酯;xlix.(S)-(R)-2-羥基-2-苯基乙酸奎寧環-3-基酯;l.(S)-(S)-2-羥基-2-苯基乙酸奎寧環-3-基酯;li.(R)-N-(1-苄基哌啶-4-基)-2-羥基-2-苯基乙醯胺;lii.(S)-N-(1-苄基哌啶-4-基)-2-羥基-2-苯基乙醯胺;liii.(R)-3-((R)-2-羥基-2-苯基乙醯氧基)-1,1-二甲基哌啶-1-鎓;liv.(S)-3-((R)-2-羥基-2-苯基乙醯氧基)-1,1-二甲基哌啶-1-鎓;lv.(R)-2-羥基-2-苯基-N-(吡啶-3-基)乙醯胺;lvi.(R)-(S)-2-乙醯氧基-2-苯基乙酸-1-甲基哌啶-3-基酯;lvii.(R)-(R)-2-羥基-2-苯基乙酸-1-甲基吡咯烷-3-基酯;lviii.(R)-(S)-2-羥基-2-苯基乙酸-1-甲基吡咯烷-3-基酯;lix.(S)-(R)-2-甲氧基-2-苯基乙酸-1-甲基哌啶-3-基酯;lx.2-甲氧基-2-苯基乙酸;lxi.(R)-2-羥基-2-苯基-N-(哌啶-4-基)乙醯胺;lxii.(R)-2-羥基-2-苯基乙酸哌啶-4-基酯;lxiii.(R)-3-(2-羥基-2,2-二苯基乙醯氧基)-1,1-二甲基哌啶-1-鎓;lxiv.(R)-3-(2-羥基-2,2-二苯基乙醯氧基)-1,1-二甲基哌啶-1-鎓;lxv.(R)-3-((R)-2-羥基-2-苯基乙醯氧基)-1,1-二甲基哌啶-1-鎓;lxvi.(R)-3-((R)-2-羥基-2-苯基乙醯氧基)-1,1-二甲基哌啶-1-鎓;lxvii.(S)-(R)-2-甲氧基-2-苯基乙酸-1-甲基哌啶-3-基酯;lxviii.(S)-(S)-2-甲氧基-2-苯基乙酸-1-甲基哌啶-3-基酯;lxix.(S)-2-羥基-N-((R)-1-甲基哌啶-3-基)-2-苯基乙醯胺;lxx.(S)-2-羥基-N-((S)-1-甲基哌啶-3-基)-2-苯基乙醯胺;lxxi.3-(2,2-二苯基乙醯胺基)-1,1-二甲基哌啶-1-鎓;lxxii.(2S)-1-苄基-2-((2,2-二苯基乙醯胺基)甲基)-1-甲基吡咯烷-1-鎓;lxxiii.3-(2-羥基-2,2-二苯基乙醯胺基)-1,1-二甲基哌啶-1-鎓;lxxiv.(R)-2-甲氧基-1-((S)-3-甲基嗎啉)-2-苯基乙酮;lxxv.(R)-2-甲氧基-1-((R)-3-甲基嗎啉)-2-苯基乙酮;lxxvi.(R)-1-((2R,3S)-2,3-二甲基嗎啉)-2-甲氧基-2-苯基乙酮;lxxvii.(S)-2-甲氧基-1-((S)-3-甲基嗎啉)-2-苯基乙酮;lxxviii.(1R,3R)-1-苄基-3-(2,2-二苯基乙醯胺基)-1-甲基哌啶-1-鎓;lxxix.(1S,3R)-1-苄基-3-(2,2-二苯基乙醯胺基)-1-甲基哌啶-1-鎓;lxxx.(1S,3R)-1-苄基-3-(2,2-二苯基乙醯胺基)-1-甲基哌啶-1-鎓;lxxxi.(1R,3R)-1-苄基-3-(2,2-二苯基乙醯胺基)-1-甲基哌啶-1-鎓;lxxxii.1-(3-(苄基氨基)哌啶-1-基)-2,2-二苯基乙酮;lxxxiii.N-((1-苄基吡咯烷-2-基)甲基)-2,2-二苯基乙醯胺;lxxxiv.1-苄基-2-((2,2-二苯基乙醯胺基)甲基)-1-甲基吡咯烷-1-鎓;lxxxv.(R)-2-甲氧基-2-苯基-N-(哌啶-4-基)乙醯胺;lxxxvi.2-羥基-2-苯基-N-(哌啶-4-基)乙醯胺;lxxxvii.(S)-2-羥基-2-苯基-N-(哌啶-4-基)乙醯胺;lxxxviii.(S)-2-甲氧基-2-苯基-N-(哌啶-4-基)乙醯胺;lxxxix.2-羥基-2-苯基-N-(哌啶-4-基)丙醯胺;xc.2-(3-溴-2,6-二氟苯基)-2-羥基-N-(哌啶-4-基)乙醯胺;xci.(R)-2-甲氧基-N-甲基-2-苯基-N-(哌啶-4-基)乙醯胺;xcii.(R)-N-(1-苄基哌啶-4-基)-2-甲氧基-N-甲基-2-苯基乙醯胺;xciii.(S)-2-甲氧基-2-苯基乙酸-1-苄基哌啶-4-基酯;xciv.(R)-4-(2-甲氧基-2-苯基乙醯胺基)哌啶-1-甲酸乙酯;B.鹽和異構體和反離子本發明在其範圍內包括鹽和異構體。本發明的化合物在新穎之後在一些情況下可以形成鹽,其也在本發明的範圍內。本發明化合物的所有立體異構體,諸如由於所述化合物的R取代基上的不對稱碳原子而可能存在的那些立體異構體,包括對映異構體和非對映異構體形式,被考慮在本發明的範圍內。本發明還可以可選地在其範圍內設想選擇合適的反離子(counterion)的效果。本發明在其範圍內包括氘代化合物的修飾。氘化化合物是其中所述化合物具有選擇性摻入代替氫的氘的那些化合物。本發明的化合物可以以其對映體純的形式或其混合物存在。C.本發明的所述化合物的合成本發明的化合物可以通過如下所示的任何合成方案來合成:常規的合成方案本發明的所述化合物可以通過如上面方案中所述的在(i)的存在下將A與B偶聯而合成。X選自O、NH或N(烷基);R1和R2獨立地選自氫、氘、羥基、C1-10直鏈或支鏈烷基、3-7元環烷基、C1-6烷氧基、芳基、氨基、NH(烷基)、N(烷基)2、OCOR5、含有1-3個選自包括O、N或S的組的雜原子的雜芳基;或者R1和R2可以被組合以形成含有1-3個選自包括O、N或S的組的雜原子的芳基或雜芳基環;R3選自氫、羥基、C1-6直鏈或支鏈烷基、3-7元環烷基、C1-6烷氧基、芳基、選自以下結構式的芳香族或非芳香族雜環或稠合雜環:X可以與R3一起形成包含1-3個選自包含N、O或S的組的雜原子的5-7元雜環。所述雜環進一步被一個或多個低級烷基、滷素、氨基、NH(烷基)、N(烷基)2、NH-芳烷基取代;R4選自氫、低級直鏈或支鏈烷基、滷素、氘、C1-6烷氧基、氨基、NH(烷基)、N(烷基)2、-COOR8、CONR8R9;R5是氫、羥基、C1-6烷基、C1-6烷氧基、氨基、NH(烷基)、N(烷基)2;R6和R7獨立地選自包括氫、C1-10烷基、COR8、-CH2OCOR8、-CH2OCONHR8R9、-COOR8、-CONR8R9、-SO2R8、芳基、芳烷基的組;R8和R9獨立地選自包括氫、或C1-6直鏈或支鏈烷基的組;n為1、2或3。以及其鹽,水合物和立體異構體。一般的程序包括酸與胺或醇(R3X)的偶聯反應。在一種方法中,酸(A)被用作起始原料,其可以通過使用諸如亞硫醯氯或草醯氯的試劑在諸如DCM的溶劑中在有或沒有少量DMF的情況下,在室溫至回流的溫度下轉化為相應的醯氯(acidchloride)而活化,然後通過該醯氯與所需的胺或醇(R3X)的反應以得到該系列的其他成員。另一種方法包括在諸如DMF的有機溶劑中在有或沒有如DMAP的有機鹼的情況下使用如碳二亞胺的酸偶聯試劑。可替換地,酸可以通過使用醇(諸如甲醇)首先轉化為相應的酯而活化,然後使其與胺或醇在溶劑(諸如苯)中在有或沒有鹼(諸如甲醇鈉)的情況下反應,以得到該系列的其他成員。D.使用的方法和含有本發明的新實體的藥物組合物因此,本發明提供了如本文所定義的新化合物用於人類或獸醫藥中的用途。本發明的化合物可用於治療和預防由分枝桿菌引起的疾病(諸如結核病)。所述結核病可由分枝桿菌種引起,所述分枝桿菌種由結核分枝桿菌(Mycobacteriumtuberculosis)、牛分枝桿菌(M.bovis)、非洲分枝桿菌(M.africanum)、坎納分枝桿菌(M.canettior)、田鼠分枝桿菌(M.microti)組成。特別地,本發明的所述化合物有效地抑制結核分枝桿菌的生長。如本文所提及的結核病包括活動性結核病或潛伏性結核病。活動性結核病包括藥物敏感性、單藥耐藥性、多藥耐藥性結核病(MDR)、廣泛耐藥性結核病(XDR)或完全耐藥性結核病。在一個方面,本發明的化合物可用於治療包括MDR、XDR和TDR結核病的結核病。在一個方面,本發明的化合物可單獨使用或與環絲氨酸、阿米卡星、利奈唑胺(linezolid)、乙胺丁醇、利福平、異煙肼、乙硫異煙胺、莫西沙星(moxyfloxacin)、克拉黴素(clarithromycin)、PAS、氯法齊明(氯苯吩嗪,clofazamine)、鏈黴素、捲曲黴素和卡那黴素和DOTS(「直接觀察治療短程」)療法組合使用。在另一方面,本發明的化合物還可以預防性地用於預防直接與結核病患者接觸的醫療保健提供者、衛生工作者、社區成員等的結核病的發生。本發明的化合物可用於治療由如上所定義的耐藥性和非耐藥性結核分枝桿菌引起的感染。本發明的所述化合物針對結核病是有效的,並且所述結核病包括由結核分枝桿菌分枝桿菌菌株中的一株所引起的藥物敏感性、單藥耐藥性、多藥耐藥性(MDR)、廣泛耐藥性(XDR)和完全耐藥性(TDR)。本發明的所述化合物可用於治療由結核分枝桿菌菌株引起的多重耐藥性(MDR)、廣泛耐藥性(XDR)和完全耐藥性(TDR)結核病,以及通過由藥劑(agent)抑制GPR109A抑制而用於治療由如上所定義的耐藥性和非耐藥性結核分枝桿菌引起的感染。用作藥物的化合物可以作為藥物組合物存在。因此,本發明在另一方面提供了包含本發明的所述新化合物連同其藥學上可接受的賦形劑/載體和可選的其他治療性和/或預防性成分的藥物組合物。所述賦形劑/載體在與所述組合物的其他成分相容的意義上必須是「可接受的」,並且對其接受者無害。合適地,該藥物組合物將在適當的製劑中。所述藥物製劑可以是任何製劑,並且包括適合於口服、鼻內或腸胃道外(包括肌內和靜脈內)施用的那些製劑。在適當的情況下,所述製劑可方便地以離散劑量單位存在,並且可通過藥學領域公知的任何方法製備。所有方法包括使所述活性化合物與液體載體或細碎的固體載體或兩者都有結合的步驟,然後如果需要,使產物成形為所需的製劑。為了這些目的,本發明的所述化合物可以以含有常規無毒的藥學上可接受的載體(carrier)、佐劑和媒介(載體,vehicle)的劑量單位製劑口服地、局部地、鼻內地、腸胃外地、通過吸入噴霧或直腸地被施用。如本文所用的術語腸胃外包括皮下注射、靜脈內、肌內、腔內注射或輸注技術。除了治療溫血動物(諸如小鼠、大鼠、馬、狗、貓等),本發明的所述化合物在人類的治療中也是有效的。在一方面,本發明的化合物可以以每天0.1至100mg/kg體重的劑量範圍給藥。本發明的所述化合物可用於預防和治療結核病,並且可被用作GPR109A抑制劑。E.實驗本發明的化合物可以通過如本文下面的方案來製備:合成方案1:步驟1:向KOH(21.0gm)的水(42.0ml)溶液中加入乙醇(54.0ml)和該化合物[1](25.0g,119mmol),並將所得的溶液回流30分鐘,並倒入玻璃板中,並在室溫下放置過夜。將所得的半固體溶於水(400ml)中,並用乙酸乙酯洗滌。用50%的HCl將水層的pH調節至酸性,並用乙酸乙酯萃取。乙酸乙酯層用無水Na2SO4乾燥並濃縮,以得到2-羥基-2,2-二苯基乙酸(12.0g,45%)。分析數據:[2]ESIMS:229[M++1]步驟2在0℃下向上述步驟中獲得的化合物(12g,52.6mmol)在甲醇(100.0ml)中的溶液中加入亞硫醯氯(5.0ml),並將所得的溶液回流4小時。將乙醇在真空下濃縮,並且殘餘物通過柱色譜使用在己烷中的20%的乙酸乙酯純化,以得到2-羥基-2,2-二苯基乙酸乙酯的液體(10.08g,76%)。分析數據:[3]ESIMS:257[M++1]步驟3:向[3](1.00g,5.23mmol)在苯(90mL)中的溶液中加入鈉(110mg)。在回流2.5小時後,加入二苯乙醇酸甲酯(1.27g,5.23mmol)的溶液,並且使反應回流過夜。在真空中蒸發苯,並且殘餘物通過快速色譜法(首先用20%的EtOAc/己烷洗脫,然後用5%的MeOH/CH2Cl2洗脫)純化,以得到作為透明液體(981mg,46%)的[1004]。分析數據[1004]ESIMS:402[M++1]步驟4:在室溫下向攪拌的[1004](0.5g,1.24mmol)在乙酸乙酯和甲醇(1:1、5ml)的混合物中的溶液中加入10%的Pd/C(0.02g)的漿料。施加氫氣球壓力並將反應混合物在室溫下攪拌3小時。使用TLC監測反應。將反應物料經硅藻土過濾,並在真空下除去過量的溶劑,以得到淺棕色的粘性物質,使用矽膠柱和在二氯甲烷中的2%的甲醇作為洗脫劑對該粘性物質進行進一步純化,以得到作為灰白色粘性物質的[1003](0.22g,57%)。分析數據:[1003]ESIMS:312[M++1]步驟5:在0℃下,在氮氣氣氛下向攪拌的[1003](0.12g,0.38mmol)在DMF中的溶液中加入無水K2CO3(0.05g,0.41mmol)。在相同的溫度下另外攪拌15分鐘後,逐滴加入碘甲烷(Methyliodide)(0.02ml,0.41mmol)。使反應溫度升至25℃,並繼續攪拌3小時。通過TLC監測[1003]的消耗。在完全消耗[1003]後,加入水(50ml),並且有機層用乙酸乙酯(2×50ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。濃縮該有機層,以得到淺棕色粘性物質,使用5%的乙酸乙酯/己烷作為洗脫劑,使用矽膠柱色譜對所述粘性物質進行進一步純化,以得到作為白色固體的[1001](0.08g,65%)。分析數據:[1001]ESIMS:326[M++1]。步驟6:在0℃下,在氮氣氣氛下向攪拌的[1003](0.05g,0.16mmol)的DCM溶液中加入三乙胺(0.03ml,0.24mmol)。在相同的溫度下另外攪拌5分鐘後,滴加二甲氨基甲醯氯(0.02ml,0.24mmol)。使反應溫度升至25℃,並繼續攪拌1小時。通過TLC監測[1003]的消耗。在[1003]完全消耗後,加入水(50ml),並且用乙酸乙酯(2×25ml)萃取有機層。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘性物質,將該粘性物質進一步用DCM和戊烷洗滌,以得到[1006](0.04g,65%)。分析數據:[1006]ESIMS:385[M++1]。[1010]、[1012]、[1008]、[1013]、[1018]、[1007]、[1009]、[1014]、[1019]和[1023]的合成通過針對[1004]所描述的程序進行。[1005]、[1011]和[1015]的合成通過針對[1006]所描述的程序進行。合成方案2:步驟1:在室溫下向攪拌的[5](2.0g,10.47mmol)的溶液中加入乙酸乙烯酯(10.0ml,100.0mmol)。在相同的溫度下另外攪拌5分鐘後,加入PS-IM(0.20g,10%)。使反應溫度升至40℃,並繼續攪拌16小時。使反應混合物穿過硅藻土床並蒸發至幹,使用10%的乙酸乙酯/己烷作為洗脫劑,使用矽膠柱色譜對其進行進一步純化,以得到作為透明粘性物質的[6](0.9g,45%)和[7](0.8g,40%)。分析數據:[6]ESIMS:192[M++1][7]ESIMS:234[M++1]。步驟2:向[6](0.45g,1.8mmol)在苯(45mL)中的溶液中加入鈉(0.03g,1.6mmol)。在回流2.5小時後,加入苯甲酸乙酯(0.35g,1.8mmol)的溶液,並將反應物回流過夜。在真空中蒸發苯,並且殘餘物通過快速色譜法(先用20%的EtOAc/己烷,然後用5%的MeOH/CH2Cl2洗脫)純化,以得到作為透明液體(0.35g,48%)的[1024]。分析數據:[1024]ESIMS:402[M++1]步驟3:向[7](0.60g,2.5mmol)在THF/MeOH/H2O(9ml)的混合物的溶液中加入氫氧化鋰(0.20g,5.0mmol)。將反應混合物在室溫下攪拌4小時。TLC顯示起始原料的完全消耗。將反應混合物蒸發至幹。加入水並用乙酸乙酯(2×50ml)萃取,以得到光透明的粘性物質[8](0.35g,73%)。分析數據:[8]ESIMS:192[M++1]步驟4:如步驟2中所述的合成[1025]。分析數據:[1025]ESIMS:402[M++1]。[1026]和[1027]的合成通過針對[1024]所描述的程序進行。合成方案3:步驟1:在室溫下向攪拌的[5](1.0g,5.2mmol)在DCM(20.0ml)的溶液中加入TEA(1.9g,15.2mmol)。在相同的溫度下另外攪拌5分鐘後,加入甲磺醯氯(0.55ml,7.8mmol)。允許所述反應溫度在該溫度下攪拌3小時。TLC顯示起始原料的完全消耗。加入水(100ml),並且有機層用乙酸乙酯(2×100ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘性物質[9],其無需任何純化被進一步使用(1.1g,78%)。分析數據:[9]ESIMS:270[M++1]。步驟2:在室溫下,向攪拌的[9](0.5g,1.8mmol)在DMF(10.0ml)中的溶液中加入疊氮化鈉(0.46g,7.2mmol)。將反應混合物的溫度升至100℃,並允許在該溫度下加熱2小時。TLC顯示起始原料的完全消耗。加入水(100ml),並且有機層用乙酸乙酯(2×100ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘性物質[10],其無需任何純化而被進一步使用(0.3g,77%)。分析數據:[10]ESIMS:217[M++1]。步驟3:在室溫下向攪拌的[10](0.3g,1.3mmol)在THF(10.0ml)中的溶液中加入三苯基膦(0.5g,1.9mmol)。在相同的溫度下另外攪拌5分鐘後,加入水(0.2ml,9.1mmol)。使反應溫度在70℃下回流2小時。TLC顯示起始原料的完全消耗。將反應混合物蒸發,並且加入水(100ml),且有機層用乙酸乙酯(2×100ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘稠物,將其用2%的MeOH/DCM作為洗脫劑進行柱色譜層析,以得到淺棕色粘性物質[11](0.2g,80%)。分析數據:[11]ESIMS:191[M++1]步驟4:在室溫下向攪拌的[12](0.2g,0.87mmol)在DMF(5.0ml)的溶液中加入EDC(0.25g,1.3mmol)、HOBT(0.17g,1.3mmol)。在相同的溫度下另外攪拌5分鐘後,加入[11](0.18g,0.91mmol)和NMM(0.37ml,2.61mmol)。將反應溫度在室溫下攪拌過夜。TLC顯示起始原料的完全消耗。加入水(100ml),並且有機層用乙酸乙酯(2×100ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘稠物,將其用1%的MeOH/DCM作為洗脫劑進行柱色譜層析,以得到淺棕色固體物質[13](0.25g,76%)。分析數據:[13]ESIMS:400[M++1]步驟5:在室溫下向攪拌的[13](0.25g,0.62mmol)在乙酸乙酯和甲醇(1:1、5ml)的混合物的溶液中加入10%的Pd/C(0.02g)的漿料。施加氫氣球壓力並將反應混合物在室溫下攪拌3小時。使用TLC監測反應。將反應物料經硅藻土過濾,並且在真空下除去過量的溶劑,以得到淺棕色粘性物質,使用矽膠柱和在作為洗脫劑的二氯甲烷中的2%的甲醇對該粘性物質進行進一步純化,以得到作為灰白色粘性物質(0.11g,57%)的[1016]。分析數據:[1016]ESIMS:311[M++1]步驟6:在氮氣氣氛下在0℃下向攪拌的[1016](0.10g,0.32mmol)在DMF的溶液中加入無水K2CO3(0.06g,0.48mmol)。在相同的溫度下另外攪拌15分鐘後,逐滴加入碘甲烷(0.03ml,0.48mmol)。使反應溫度升至25℃,並繼續攪拌3小時。通過TLC監測[1016]的消耗。在完全消耗[1016]後,加入水(50ml),並且有機層用乙酸乙酯(2×50ml)萃取。合併的有機層用水、鹽水洗滌,並且用硫酸鈉乾燥。濃縮有機層,以得到淺棕色粘性物質,使用10%的乙酸乙酯/己烷作為洗脫劑,使用矽膠柱色譜對該粘性物質進行進一步純化,以得到作為白色固體的[1020](0.05,50%)。分析數據:[1020]ESIMS:325[M++1][1017]、[1021]、[1022]、[1028]、[1029]、[1030]、[1031]和[1032]的合成通過針對[1020]所描述的程序進行。合成方案4:步驟1:在室溫下,在氮氣氣氛下向[14](0.50g,4.34mmol)在苯(30ml)的攪拌溶液中加入PTSA(0.74g,4.34mmol)。在相同的溫度下另外攪拌15分鐘後,加入[15](0.65g,4.34mmol)。允許反應溫度升高至90℃,並繼續攪拌5小時。通過TLC監測[15]的消耗。在[15]完全消耗後,加入水(50ml),並且有機層用乙酸乙酯(3×50ml)萃取。合併的有機層用水、鹽水洗滌,並且用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘性物質,使用30%的乙酸乙酯/己烷作為洗脫劑,使用矽膠柱色譜對粘性物質進行進一步純化,以得到[1037]和[1038]的混合物,其通過製備性HPLC被進一步分離,以得到作為透明粘性物質的[1037](0.10g,9%)和[1038](0.08g,8%)。分析數據:[1037和1038]ESIMS:250[M++1]。步驟2:在室溫下,在氮氣氣氛下向[1037](0.05g,0.20mmol)在乙腈(2ml)的攪拌溶液中加入碘甲烷(0.01ml,0.20mmol)。將反應溫度在該溫度下攪拌過夜。在完全消耗[1037]後,將反應混合物蒸發至幹,以得到淺黃色固體,通過用二氯甲烷和乙醚洗滌而將該黃色固體進一步純化,以得到純的產物[1065](0.04g,51%)。分析數據:[1065]ESIMS:364[M+]。步驟3:類似於如步驟2中所描述的[1065]來合成[1064]。分析數據:[1064]ESIMS:364[M+]。[1039]、[1040]、[1041]、[1042]、[1043]、[1044]、[1045]、[1046]、[1047]、[1048]、[1049]、[1050]、[1051]、[1052]、[1055]、[1057]、[1058]、[1059]、[1053]、[1054]、[1063]、[1067]、[1068]、[1069]、[1070]、[1071]、[1073]、[1061]、[1062]、[1074]、[1075]、[1076]、[1077]和[1093]的合成通過針對[1064]和[1065]所描述的程序進行。合成方案5:步驟1:在室溫下向[17](0.08g,0.42mmol)在DMF(5.0ml)中的攪拌溶液中加入EDC(0.12g,0.63mmol)、HOBT(0.08g,0.63mmol)。在相同的溫度下另外攪拌5分鐘後,加入[16](0.08g,0.42mmol)和NMM(0.14ml,2.3mmol)。允許反應溫度在室溫下攪拌過夜。TLC顯示起始原料的完全消耗。加入水(100ml),並且有機層用乙酸乙酯(2×100ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘稠物,將其用作為洗脫劑的1%的MeOH/DCM進行柱色譜層析,以得到白色固體物質[18](0.11g,68%)。分析數據:[18]ESIMS:385[M++1]。步驟2:在室溫下,在氮氣氣氛下向[18](0.11g,0.28mmol)在乙腈(2ml)的攪拌溶液中加入碘甲烷(0.02ml,0.28mmol)。將反應溫度在該溫度下攪拌過夜。在完全消耗[18]後,將反應混合物蒸發至幹,以得到作為[11081]和[11082]的混合物的淺黃色固體,其通過製備性HPLC進一步純化,以得到作為淺黃色固體物質的[1078](0.03g,20%)和[1079](0.4g,27%)。分析數據:[1078和1079]ESIMS:399[M+]。[1080]、[1081]、[1082]、[1083]、[1084]和[1034]的合成通過針對[1078]和[1079]所描述的程序進行。合成方案6:步驟1:在室溫下向[19](0.80g,4.81mmol)在DMF(5.0ml)中的攪拌溶液中加入EDC(1.40g,7.2mmol)、HOBT(0.97g,7.2mmol)。在相同的溫度下另外攪拌5分鐘後,加入[20](1.11ml,5.30mmol)和NMM(1.6ml,14.43mmol)。允許反應溫度在室溫下攪拌過夜。TLC顯示起始原料的完全消耗。加入水(100ml),並且有機層用乙酸乙酯(2×100ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺黃色粉末,將其用戊烷研磨,以得到灰白色固體物質[21](1.20g,75%)。分析數據:[21]ESIMS:339[M++1]。步驟2:在室溫下向[21](0.5g,1.4mmol)在甲醇(10ml)中的攪拌溶液中加入10%的Pd/C(0.05g)的漿料。施加氫氣球壓力並將反應混合物在室溫下攪拌3小時。使用TLC監測反應。反應物料經硅藻土過濾,並且在真空下除去過量的溶劑,以得到灰白色粉末[1085](0.3g,86%)。分析數據:[1085]ESIMS:249[M++1]。步驟3:在0℃下,在氮氣氣氛下向[1085鹽酸鹽](0.05g,0.20mmol)在DCM中的攪拌溶液中加入三乙胺(0.07ml,0.5mmol)。在相同的溫度下另外攪拌5分鐘後,逐滴加入氯甲酸乙酯(0.02ml,0.22mmol)。允許反應溫度升至25℃,並繼續攪拌1小時。通過TLC監測[1085]的消耗。在完全消耗[1085]後,加入水(50ml),並且有機層用乙酸乙酯(2×25ml)萃取。合併的有機層用水、鹽水洗滌,並用硫酸鈉乾燥。將有機層濃縮,以得到淺棕色粘性物質,其進一步用DCM和戊烷洗,以得到[1094](0.03g,64%)。分析數據:[1094]ESIMS:321[M++1]。[1086]、[1087]、[1088]、[1089]、[1090]、[1091]、[1092]、[1035]和[1036]的合成通過針對[1085]所描述的程序進行。F.本發明化合物的生物測試實施例1:本發明的化合物對巨噬細胞中分枝桿菌菌落生長的抑制。將THP1細胞接種在完全RPMI+10%的FCS的組織培養板中,並且通過加入PMA伴隨在37℃下用5%的CO2下孵育,使THP1細胞分化成巨噬細胞。在PMA分化16-20小時後,洗滌細胞並用完全RPMI補充。用結核分枝桿菌細菌H37Rv以10的MOI感染PMA分化的THP-1細胞。在補充有10%的FCS的無抗生素的RPMI中進行感染。在加入細菌後,將培養板離心,然後在37℃下用5%的CO2下孵育。4小時後,用溫熱的RPMI洗滌感染的細胞兩次,並補充含有阿米卡星的完全RPMI以除去任何剩餘的細胞外細菌。在16小時時加入濃度為5至500nM的化合物,並且每24小時至64小時的時間點,將含有適當劑量的化合物的培養基更新。在90小時的測定結束時,將細胞用裂解緩衝液(7H9+.06%的SDS)裂解,並將殘餘的細菌負荷確定為CFU計數。結果以圖表的方式示於表2中,其表明一旦使用這些化合物後分枝桿菌菌落的生長受到抑制。表2:在100nM和500nM下篩選化合物對H37Rv的抑制%NT=未測試實施例3:人PBMC衍生的巨噬細胞的體外感染、化合物添加和cfu測定。用RPMI1640以1:1稀釋的肝素化人血液被成層堆積到等體積的Ficoll-Paque上,隨後以1600rpm離心30分鐘。小心地收集在界面處形成的PBMC層,並用RPMI洗滌兩次。將所述細胞在RMPI培養基(無血清)中稀釋至2×106/ml的濃度,並將10ml的稀釋的細胞置於75cm2的組織培養瓶中,並在加溼的37℃培養箱中孵育2小時。通過抽吸除去非貼壁細胞,隨後用RPMI洗滌兩次。加入完全培養基(含10%的FCS),並且允許細胞在加溼的37℃、5%的CO2培養箱中自發分化成巨噬細胞4天。細菌在補充有10%的ADC、0.4%的甘油和0.05%的吐溫-80的Middlebrooke7H9肉湯中生長直至對數中期。然後收穫所述細菌,用RPMI洗滌並重新懸浮在相同的培養基中。通過每次用23號針頭然後用26號針頭抽吸12次來分散所述懸浮液,接著通過30號針頭進行額外的分散3次。將該懸浮液靜置5分鐘。然後將懸浮液的上半部分用於實驗。通過測量在600nm波長處的吸光度(0.6O.D.對應於約100×106個細菌)來定量細菌。以10的MOI(即,每個細胞10個細菌)用結核分枝桿菌感染人PBMC衍生的巨噬細胞。在補充有10%的FCS的無抗生素的RPMI中進行感染。在加入細菌後,在37℃下用5%的CO2孵育之前,將培養板以700rpm離心5分鐘。4小時後,用溫熱的RPMI洗滌感染的細胞兩次,並用含有200μg/mL阿米卡星的完全RPMI補充2小時以除去任何剩餘的細胞外細菌。隨後,洗滌細胞,然後將所述細胞保持在完全RPMI中用於實驗的其餘部分。在感染後(p.i)16小時進行抑制劑的添加,並且在感染後(p.i)40小時和64小時重新補充含有合適劑量的抑制劑的培養基。在感染後90小時,將所述細胞在50μl的0.06%的SDS中在室溫下裂解10分鐘。將1:10的裂解物稀釋物一式兩份鋪板於7H11瓊脂板上。通過追蹤稀釋法(軌道稀釋法)使用方形板(12×12cm)進行鋪板,其中將10μl的每種稀釋物點在方形板的一側上。然後將該板傾斜在其側面(以45°-90°的角度)上,並使所述斑點沿著瓊脂表面在平行的軌道中輕輕流動。然後使該板乾燥,並隨後在加溼的培養箱中在37℃下孵育。在第14天對菌落進行計數並轉換為cfu/孔。本發明的化合物中的一種在減少人PBMC衍生的巨噬細胞中生長的分枝桿菌的效果作為說明的手段(方式)示於表3中。表3:使用化合物1085時菌落計數減少的證明實施例4:THP-1巨噬細胞的體外感染、化合物添加和cfu測定。所述人單核細胞/巨噬細胞細胞系THP-1在補充有10%的FCS的RPMI1640中培養,並在37℃下在加溼的5%的CO2氣氛中保持在2×105到10×105個細胞/ml之間。在感染前,將細胞以1×104個細胞/孔鋪在96孔板中,並用PMA(30ng/ml)分化48小時的時間。細菌在補充有10%的ADC、0.4%的甘油和0.05%的吐溫80的Middlebrooke7H9肉湯中生長直到對數中期。然後收穫所述細菌,用RPMI洗滌並重新懸浮於相同的培養基中。通過每次用23號針頭以及然後用26號針頭抽吸12次來分散所述懸浮液,隨後通過30號針頭額外分散3次。將該懸浮液靜置5分鐘。然後將懸浮液的上半部分用於實驗。通過測量在600nm波長處的吸光度(0.6O.D.對應於約100×106個細菌)來定量細菌。以10的MOI(即,每個細胞10個細菌)用結核分枝桿菌感染PMA衍生的THP-1細胞。在補充有10%的FCS的無抗生素的RPMI中進行感染。在加入細菌後,在37℃下用5%的CO2下孵育之前,將培養板以700rpm離心5分鐘。4小時後,用溫熱的RPMI洗滌感染的細胞兩次,並用含有200μg/mL阿米卡星的完全RPMI補充2小時以除去任何剩餘的細胞外細菌。隨後,洗滌細胞,然後將所述細胞保持在完全RPMI中用於實驗的其餘部分。在感染後(p.i)16小時進行抑制劑的添加,並且在感染後(p.i)40小時和64小時補充含有合適劑量抑制劑的培養基。在感染後90小時,將所述細胞在50μl的0.06%的SDS中在室溫下裂解10分鐘。將1:10的裂解物稀釋物一式兩份鋪板於7H11瓊脂板上。通過追蹤稀釋法使用方形板(12×12cm)進行鋪板,其中將10μl的每種稀釋物點在方形板的一側上。然後將該板傾斜在其側面(以45°-90°的角度)上,並使所述斑點沿著瓊脂表面在平行的軌道中輕輕流動。然後使該板乾燥,隨後在加溼的培養箱中在37℃下孵育。在第14天對菌落進行計數並轉換為cfu/孔。該結果示於圖1。從圖1可以推斷,本發明的化合物在MYC431感染的巨噬細胞中產生分枝桿菌cfu的劑量依賴性減少。實施例5:本發明的化合物對脂質體的作用的測定。將THP-1細胞以每個蓋玻片0.3×106個細胞的密度接種在24孔組織培養板中的1號厚度、12mm直徑的玻璃蓋片上。然後將這些細胞以10的MOI用M.tb(H37Rv)感染,並在37℃下在5%的CO2中孵育4小時。通過洗滌除去細胞外細菌,並隨後用200μg/ml的阿米卡星補充培養基2小時。在阿米卡星處理後立即以增加的濃度加入SPR113,並且在感染後16小時和40小時補充含有合適抑制劑的培養基。在感染後48小時,用3.7%的多聚甲醛固定細胞並用PBS洗滌。將在PBS中1:1000稀釋的HCSLipidToxRed中性脂質染料加入到細胞中持續30分鐘。使用300nM的DAPI溶液(在H2O中)將細胞核染色5分鐘,然後洗滌。用配備有60X/1.4NAPlanApochromatDIC物鏡的NikonEclipseTi-E雷射掃描共聚焦顯微鏡來觀察染色的細胞。DAPI和脂質Tox分別用藍色二極體和氦-氖雷射器在408nm和543nm激發。通過設置在450和605/75nm的發射濾光片記錄該發射。用512×512像素的掃描模式格式獲取圖像。將傳輸和檢測器增益被設置為實現最佳信噪比,並且調整雷射器功率以限制漂白螢光。使用Image-ProPlus版本6.0(一種可從MediaCybernetics商購的軟體包)來定量所有圖像。結果示於下面的圖2。圖2顯示了在本發明的化合物存在下細胞脂質體的劑量依賴性減少。實施例6:化合物對不同的分枝桿菌菌株的影響。用rM.tb菌株[JAL2287、JAL2261、XDR]獨立地感染THP-1細胞。在16小時時加入含有化合物的培養基,並且每24小時至64小時時間點,更新含有適當劑量的化合物的培養基。在90小時的總培養期結束時,裂解細胞並測定CFU,且結果如圖3中圖表所示。在MTB感染的THP1中測量了化合物1085的細胞效能,且H37Rv的EC50為0.5nM,以及MYC431和JAL2287的EC50為0.1nM。該圖顯示化合物在0.01至500nM的濃度下抑制了所有分枝桿菌菌株。實施例7:化合物對c-AMP水平的影響。將THP-1巨噬細胞與處於所顯示濃度的化合物1085一起孵育30分鐘,然後將3-羥基丁酸(3HBA,10uM)(一種GPR109A激動劑)加入到細胞中持續90分鐘。在不存在化合物1085的情況下,3HBA降低了細胞內的cAMP水平,而化合物1085以劑量依賴性的方式抑制了該活性。該結果示於圖4中。實施例8:結核分枝桿菌與降解性囊泡的共定位:在化合物1085存在下酸化的溶酶體和自噬體將THP-1細胞以每個蓋玻片0.3×106個細胞的密度接種在24孔組織培養板中的玻璃蓋玻片上。然後將這些細胞以10的MOI感染GFP-標記的M.tb(H37Rv-GFP),並在37℃下在5%的CO2中孵育4小時。通過洗滌並隨後通過用200μg/ml的阿米卡星補充培養基2小時來除去細胞外細菌。在阿米卡星處理後立即以100nM的濃度加入化合物1085,並且在感染後16小時和40小時補充含有該化合物的培養基。在48小時,將細胞與100nM的嗜酸性染料一起孵育60分鐘,隨後用3.7%的多聚甲醛固定細胞20分鐘,並洗滌。染料染色的細胞用0.2%(v/v)的TritonX-100透化20分鐘,用PBS洗滌,並加入封閉緩衝液3%(w/v)的BSA持續60分鐘。洗滌細胞,並在室溫下加入1:200稀釋的LC3B兔Ab(細胞信號轉導技術)持續60分鐘。用PBST(一次)和PBS(兩次)洗滌細胞。在室溫下加入1:200稀釋的AlexaFluor405山羊抗兔抗體(Ab)持續45分鐘。用PBST(一次)和PBS(兩次)洗滌細胞。將蓋玻片用防褪色試劑固定在載玻片上。用雷射掃描共聚焦顯微鏡觀察染色的細胞。GFP和染料分別用氬離子、藍色二極體和氦-氖雷射器在488nm、408nm和543nm激發。通過設置在515/30nm、450nm和605/75nm的發射濾光片記錄該發射。在單個通道(綠色、藍色和紅色)中,使用電動機驅動聚焦系統獲取跨越10μm總厚度的z層疊內的系列共聚焦切片(0.5μm厚)。使用512×512像素的掃描模式格式獲取圖像。將傳輸和檢測器增益設置為實現最佳信噪比,並且調整雷射器功率以限制漂白螢光。對所有圖像進行定量。將合併的共聚焦圖像解卷積並進行共定位分析以確定如先前所述的「重疊係數」(Manders等人,1993)。結果示於圖5a和圖5b中。自噬體和酸化的溶酶體是在維持細胞內穩態方面起作用的降解性囊泡,並且也是由巨噬細胞引起的以清除細胞內感染的抗微生物應答的重要成分。結核分枝桿菌調節這些降解途徑以便確保其在巨噬細胞內的持續存活。導致在感染的巨噬細胞中這些途徑活化的幹預將導致細胞內結核分枝桿菌的靶向殺傷,並因此降低分枝桿菌負荷。用化合物1085進行的實驗的重要性在於,用該化合物處理感染的巨噬細胞導致分枝桿菌與這些降解性囊泡的共定位的增加,其是由共定位係數的增加值反映的(更高的共定位係數值表明在這些降解性囊泡內存在更高百分比的細菌)。因此,在化合物1085處理後存在於這些囊泡內部的細菌準備好被隨後殺死,其反過來被反映在利用化合物1085獲得的更低的體外cfu中。實施例9:化合物編號1085對細胞脂質體的GPR109A特異性效應。將THP-1細胞以每個蓋玻片0.3×106個細胞的密度接種在24孔組織培養板中的玻璃蓋玻片上。將GPR109A的配體,3-HB(10μM)加入到細胞和平行組中。化合物1085以增加的濃度被加入。在3HB添加後48小時,用3.7%的多聚甲醛固定細胞並用PBS洗滌。將在PBS中1:1000稀釋的HCS脂質Tox紅中性脂質染料加入細胞中30分鐘。使用300nMDAPI溶液(在H2O中)將細胞核染色5分鐘,然後洗滌。用配備有ApochromatDIC物鏡的雷射掃描共聚焦顯微鏡觀察染色的細胞。DAPI和脂質Tox分別用藍色二極體和氦-氖雷射在408nm和543nm激發。通過設置在450nm和605/75nm的發射濾光片記錄發射。使用512×512像素的掃描模式格式獲取圖像。將傳輸和檢測器增益設置為實現最佳信噪比,並且調整雷射功率以限制漂白螢光。對所有圖像進行定量,並且結果在圖6中圖示出,其證明了在化合物1085的存在下,觀察到了對代表細胞內脂質體減少的平均螢光強度的顯著影響。實施例10:化合物1085對分別感染8種不同的結核分枝桿菌菌株的THP-1巨噬細胞中細胞內分枝桿菌負載的效應人單核細胞/巨噬細胞細胞系THP-1在補充有10%的FCS的RPMI1640中培養,並在37℃下在溼潤的5%的CO2氣氛中保持在2×105和10×105個細胞/ml之間。在感染前,將細胞以1×104個細胞/孔鋪在96孔板中,並用PMA(30ng/ml)分化48小時的時間。細菌在補充有10%的ADC(BectonDickinson)、0.4%的甘油和0.05%的吐溫80的Middlebrooke7H9肉湯中生長直到對數中期。然後收穫細菌,用RPMI洗滌並重懸浮在相同的培養基中。通過每次用23號和隨後的26號針頭抽吸12次來分散懸浮液,然後通過30號針頭另外分散3次。將該懸浮液靜置5分鐘。然後將懸浮液的上半部分用於實驗。通過測定在600nm波長處的吸光度(0.6O.D.對應於約100×106個細菌)來定量細菌。以10的MOI(即,每個細胞10個細菌)用結核分枝桿菌感染PMA衍生的THP-1細胞。在補充有10%的FCS的無抗生素的RPMI中進行感染。在加入細菌後,在37℃下用5%的CO2孵育之前,將培養板以700rpm離心5分鐘。4小時後,用溫熱的RPMI洗滌感染的細胞兩次,並用含有200μg/mL的阿米卡星的完全RPMI補充2小時以除去任何剩餘的細胞外細菌。隨後,洗滌細胞,然後將所述細胞保持在完全RPMI中用於實驗的其餘部分。在感染後(p.i)16小時進行抑制劑的添加,並且在感染後(p.i)40小時和64小時補充含有合適劑量抑制劑的培養基。在感染後90小時,將所述細胞在50μl的0.06%的SDS中在室溫下裂解10分鐘。將1:10的裂解物稀釋物一式兩份鋪板於7H11瓊脂板上。通過追蹤稀釋法使用方形板(12×12cm)進行鋪板,其中將10μl的每種稀釋物點在方形板的一側上。然後將該板傾斜在其側面(以45°-90°的角度)上,並使所述斑點沿著瓊脂表面在平行的軌道中輕輕流動。然後使該板乾燥,隨後在加溼的培養箱中在37℃下孵育。在第14天對菌落進行計數並轉換為cfu/孔。該結果圖示於圖7中,圖7表示了對於隨著用於所有8種結核分枝桿菌菌株的化合物1085的濃度的提高的分枝桿菌負載的穩定降低。實施例11:化合物1085與已知抗分枝桿菌抗生素的組合對感染有結核分枝桿菌H37Rv的THP-1巨噬細胞中的細胞內分枝桿菌負載的效應。人單核細胞/巨噬細胞細胞系THP-1在補充有10%的FCS(Hyclone)的RPMI1640(Gibco實驗室公司)中培養,並在37℃下在溼潤的5%的CO2氣氛中保持在2×105到10×105個細胞/ml之間。在感染前,將細胞以1×104個細胞/孔鋪在96孔板中,並用PMA(30ng/ml)分化48小時的時間。細菌在補充有10%的ADC(BectonDickinson)、0.4%的甘油和0.05%的吐溫80的Middlebrooke7H9肉湯(Difco)中生長直到對數中期。然後收穫細菌,用RPMI洗滌並重懸浮在相同的培養基中。通過每次用23號和隨後的26號針頭抽吸12次來分散懸浮液,然後通過30號針頭另外分散3次。將該懸浮液靜置5分鐘。然後將懸浮液的上半部分用於實驗。通過測定在600nm波長處的吸光度(0.6O.D.對應於約100×106個細菌)來定量細菌。以10的MOI(即,每個細胞10個細菌)用結核分枝桿菌感染PMA衍生的THP-1細胞。在補充有10%的FCS的無抗生素的RPMI中進行感染。加入細菌後,在37℃下用5%的CO2下孵育之前,將培養板以700rpm離心5分鐘。4小時後,用溫熱的RPMI洗滌感染的細胞兩次,並用含有200μg/mL阿米卡星的完全RPMI補充2小時以除去任何剩餘的細胞外細菌。隨後,洗滌細胞,然後將所述細胞保持在完全RPMI中用於實驗的其餘部分。在感染後(p.i)16小時進行抑制劑的添加,並且在感染後(p.i)40小時和64小時補充含有合適劑量抑制劑的培養基。在感染後90小時,將所述細胞在50μl的0.06%的SDS中在室溫下裂解10分鐘。將1:10的裂解物稀釋物一式兩份鋪板於7H11瓊脂板上。通過追蹤稀釋法使用方形板(12×12cm)進行鋪板,其中將10μl的每種稀釋物點在方形板的一側上。然後將該板傾斜在其側面(以45°-90°的角度)上,並使所述斑點沿著瓊脂表面在平行的軌道中輕輕流動。然後使該板乾燥,隨後在加溼的培養箱中在37℃下孵育。在第14天對菌落進行計數並轉換為cfu/孔。圖8顯示了該化合物1085不會不利地影響該已知的抗TB抗生素的活性,並且在一些情況下可能存在加成/協同效應。實施例12:在小鼠中化合物1085的治療(口服給藥)通過氣霧劑途徑,通過在暴露30分鐘期間遞送150-200個細菌/肺,用MDR-M.tb(JAL2287)感染幼小鼠組(4-6周齡的雌性BALB/c小鼠以5隻/組)。24小時後處死一組小鼠,並將肺勻漿塗布在7H11瓊脂平板上用於確認感染。在感染後15天,通過口服遞送該化合物來開始化合物1085處理(治療)。以下列劑量處理各組小鼠:100、30和10mg/kg體重/天(1085被溶解在PEG400中)。在處理後14天,處死小鼠,並使用勻漿器使肺和脾富集。將連續稀釋的勻漿(10-2和10-3)的等分試樣(100μl)塗布在7H11瓊脂平板上,用於測定分支桿菌負荷。在瓊脂平板上鋪板後18天計數cfu。通過腹膜內途徑的藥代動力學參數的結果圖示於圖9a和圖9b。通過口服途徑的藥代動力學參數在下面的表3和4中呈現。從圖和表可以推斷,由示例性化合物1085說明的本發明的化合物被發現在所有實驗劑量下都是有效的。化合物1085的藥代動力學(PK)數據表示在表5和表6中,表明化合物1085在小鼠和犬中都具有非常好的生物利用度。表3:肺中CFU的降低(口服給藥)化合物1085(mg/kg/天)CFU/肺(x105)降低%p值038.61021.843.50.00013014.562.40.00011007.880.00.0001表4:脾中CFU的降低(口服給藥)化合物1085(mg/kg/天)CFU/肺(x105)降低%p值011.8100.496.60.0001300.298.30.00011000.298.30.0001表5:小鼠中化合物1085的PK數據小鼠的生物利用度:47%表6:犬中化合物1085的PK數據犬生物利用度:57%實施例13:小鼠中化合物1085+ATT組合治療(口服給藥)通過氣霧劑途徑,通過在暴露30分鐘期間遞送150-200個細菌/肺,用藥物敏感的M.tb(H37Rv)感染幼小鼠組(4-6周齡的雌性BALB/c小鼠以5隻/組)。24小時後處死一組小鼠,並且將肺勻漿塗布在7H11瓊脂平板上用於確認感染。在感染後15天,以單劑量每天來開始口服藥物的處理(治療)。一組接受抗TB治療,ATT(異煙肼:1.5mg/kg+利福平:1mg/kg+吡嗪醯胺:1.5mg/kg+乙胺丁醇:1.5mg/kg),一組接受化合物1085(10mg/kg),以及平行組接受化合物1085和ATT的組合(異煙肼:1.5mg/kg+利福平:1mg/kg+吡嗪醯胺:1.5mg/kg+乙胺丁醇:1.5mg/kg+化合物1085:10mg/kg)。在治療後1周、2周和4周,將小鼠與對照(mocka治療的)組一起處死,並使用勻漿器富集肺。將連續稀釋的勻漿(10-2和10-3)的等分試樣(100μl)塗布在7H11瓊脂平板上,用於測定分支桿菌負荷。在瓊脂平板上鋪板後18天計數CFU。化合物1085與ATT的組合治療顯示了肺CFU的顯著減少,如圖10中所示。該數據表示在下面的表7中。表7:化合物1085與ATT的組合治療當前第1頁1 2 3