使用腺病毒的治療方法與流程
2023-07-11 13:35:32 1
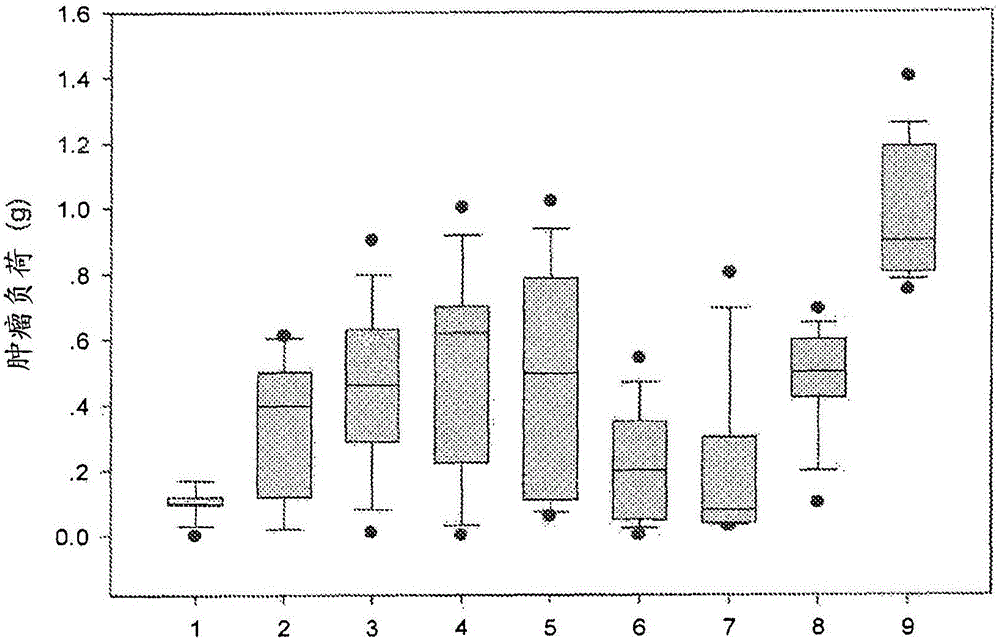
使用腺病毒的治療方法發明領域本公開內容涉及癌症生物學、免疫學和藥理學。更具體地,它涉及通過施用表達Fas嵌合轉基因產物的核酸構建體或所述核酸構建體的均一群體治療婦科癌症有關的疾病或紊亂的方法。發明背景婦科癌症在臨床上具有侵襲性,通常在女性生殖道的組織中發展,並與不良結果相關。這些癌症包括卵巢癌、子宮癌、輸卵管癌和宮頸癌,並且還包括惡性米勒管混合瘤(malignantmixed米勒管tumors,MMMT)。在極少數情況下,MMMT也能夠在女性腹膜(腹壁內膜)中發展。婦科癌症可能難於檢測並往往在它們處於晚期時才得到診斷。卵巢癌大約佔女性癌症的百分之三。作為女性中第九位最常見的癌症,卵巢癌是女性癌症相關死亡的第五大原因,並且是最致命的婦科癌症(2012年)。卵巢癌以對基於鉑和紫杉烷的療法的高反應率而對化療敏感。但是,儘管在治療的設計和遞送中的進步,癌症復發和化療抗性仍然是治療這些類型的癌症的障礙。儘管積極的主要治療和高的初始反應率,大多數患有晚期卵巢癌的女性會復發並發展抗性疾病。在這些晚期疾病狀態中,對後續化療的反應率大大減少了,突出表明迫切需要開發改進的治療劑和策略。發明概述本發明涉及降低或減少患者中腫瘤的尺寸或者消除或減緩腫瘤的生長的方法,該方法包括給所述患者施用有效量的核酸構建體,所述核酸構建體包含被可操作地連接至內皮細胞特異性啟動子的Fas嵌合基因,其中在所述患者中,由所述核酸構建體編碼的Fas嵌合基因產物降低或減少所述腫瘤的尺寸或消除所述腫瘤,並且其中所述腫瘤與女性婦科癌症或其轉移有關。本發明還提供抑制、降低或減少腫瘤中新生血管形成或血管發生的方法,該方法包括給患有所述腫瘤的患者施用有效量的核酸構建體,所述核酸構建體包含被可操作地連接至內皮細胞特異性啟動子的Fas嵌合基因,其中由所述核酸構建體編碼的Fas嵌合基因產物抑制、降低或減少所述腫瘤中新生血管形成或血管生成,並且其中所述腫瘤與女性婦科癌症或其轉移有關。此外,本發明包括治療或預防患者中與以下癌症有關或源自以下癌症的腫瘤的方法:米勒管癌症(Mülleriancancer)、卵巢癌、腹膜癌、輸卵管癌或子宮乳頭狀漿液性癌,該方法包括施用有效量的核酸構建體,所述核酸構建體包含被可操作地連接至內皮細胞特異性啟動子的Fas嵌合基因,其中由所述核酸構建體編碼的Fas嵌合基因產物治療或預防女性婦科癌症或其轉移。在一個實施方案中,所述腫瘤或其轉移在所述施用之後被降低尺寸或被消除或降低所述腫瘤的生長。在另一個實施方案中,所述腫瘤或其轉移被降低,使得與所述施用之前所述腫瘤的最大直徑(LD)相比,其LD被降低至少大約10%、20%、30%、40%、50%、60%、70%、80%、90%或100%。在其它實施方案中,所述女性婦科癌症與以下癌症有關或源自所述癌症:米勒管癌症、卵巢癌、腹膜癌、輸卵管癌、子宮乳頭狀漿液性癌、其任意組合或其轉移。在一些實施方案中,所述患者之前已經接受基於鉑的療法。在一個實例中,所述患者患有復發性鉑抗性癌症。在另一個實例中,患有復發性鉑抗性癌症或復發性紫杉烷抗性癌症的患者在完成或接受基於鉑的療法或基於紫杉烷的療法的六個月內具有進展性(progressive)腫瘤。在某些實施方案中,所述患者沒有針對腺病毒的已有抗體或沒有發展針對腺病毒的抗體。在其它實施方案中,本發明的方法還包括施用有效量的一種或更多種化療劑。所述一種或更多種化療劑可以在施用所述核酸構建體之前、同時或之後進行施用。在一個具體的實施方案中,所述化療劑是紫杉醇。在某些實施方案中,用於本發明方法的核酸構建體是腺病毒。在一個實例中,所述腺病毒表達Fas嵌合基因產物,所述Fas嵌合基因產物包含融合至Fas多肽的跨膜結構域和胞內結構域的TNF受體1(TNFR1)多肽的胞外結構域。編碼Fas嵌合基因產物的多核苷酸被可操作地連接至內皮細胞特異性啟動子,例如PPE-1-3X啟動子。在一個具體的實施方案中,所述腺病毒包含與SEQIDNO:19至少70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列。本發明的一個方面包括包含SEQIDNO:18的核酸構建體。在另一個方面,所述核酸構建體還包含編碼Fas嵌合蛋白的核苷酸序列。在其它實施方案中,本發明是包含SEQIDNO:19的載體。所述載體可以是腺病毒。在又一些其它實施方案中,本發明是具有歐洲細胞培養物保藏中心(ECACC)保藏號13021201的腺病毒。所述腺病毒可以為至少60%、至少70%、至少80%、至少90%、至少95%、至少96%、至少97%、至少98%、至少99%或至少100%純。在一個實例中,本發明包含藥物組合物,所述藥物組合物包含所述腺病毒和藥學可接受的載體,其中所述組合物不含有另一類型的腺病毒,例如包含SEQIDNO:20或SEQIDNO:21的腺病毒。本發明還包括在需要其的受試者的組織中抑制、降低或減少血管發生或新生血管形成的方法,該方法包括向所述受試者施用所述核酸構建體、所述載體、所述腺病毒或所述組合物。在一個實施方案中,所述組織包含腫瘤。在另一個實施方案中,在所述施用之後腫瘤的尺寸被減少或降低或者在所述施用之後所述腫瘤的生長被減緩。在其它實施方案中,所述腫瘤源自以下癌症或與以下癌症有關:甲狀腺癌、神經內分泌癌、膠質母細胞瘤、女性婦科癌症、其任意組合或其轉移。附圖簡要說明圖1顯示了在施用媒介物(vehicle)、VB1111E9(109個病毒顆粒(VP)的VB-111)、VB1111E11(1011個VP的VB-111)、低Com(ComLow)(20mg/kg卡鉑+10mg/kg愛寧達(Alimta))、高Com(ComHigh)(50mg/kg卡鉑+30mg/kg愛寧達)、VB1111E9+低Com(VB-111109個VP+20mg/kg卡鉑+10mg/kg愛寧達)、VB1111E9+高Com(VB-111109個VP+50mg/kg卡鉑+30mg/kg愛寧達)、VB1111E11+低Com(VB-1111011個VP+20mg/kg卡鉑+10mg/kg愛寧達)和VB1111E11+高Com(VB-1111011個VP+50mg/kg卡鉑+30mg/kg愛寧達)(x軸)的Lewis肺癌小鼠中的平均腫瘤負荷(y軸)。圖2顯示了在施用媒介物(1)、VB1111E9(109個病毒顆粒(VP)的VB-111)(2)、VB1111E11(1011個VP的VB-111)(3)、低Com(20mg/kg卡鉑+10mg/kg愛寧達)(4)、高Com(50mg/kg卡鉑+30mg/kg愛寧達)(5)、VB1111E9+低Com(VB-111109個VP+20mg/kg卡鉑+10mg/kg愛寧達)(6)、VB1111E9+高Com(VB-111109個VP+50mg/kg卡鉑+30mg/kg愛寧達)(7)、VB1111E11+低Com(VB-1111011個VP+20mg/kg卡鉑+10mg/kg愛寧達)(8)和VB1111E11+高Com(VB-1111011個VP+50mg/kg卡鉑+30mg/kg愛寧達)(9)(x軸)的Lewis肺癌小鼠中的平均腫瘤負荷(y軸)的盒型圖(boxplot)。圖3顯示了包含被可操作地連接至內皮細胞特異性啟動子(例如VB-111)的FAS嵌合基因的腺病毒和紫杉醇的組合治療方案。每八周施用大約3x1012個VP或1x1013個VP的VB-111,並且每周施用紫杉醇一次。發明詳述一、定義除非另有定義,否則本文使用的所有技術和科學術語具有如本發明所屬技術領域的普通技術人員通常理解的相同的含義。在有衝突的情況下,以本發明(包括定義)為準。除非上下文另有要求,否則單數術語應包括複數,且複數術語應包括單數。本文所提到的所有出版物、專利和其它文獻通過引用整體併入本文用於所有目的,如同每篇出版物或專利申請被具體地且個別地指明通過引用併入本文一樣。雖然與本文所述的那些類似或等同的方法和材料能夠被用於實踐或測試本發明,合適的方法和材料被描述如下。所述材料、方法和實例僅僅是說明性的而非旨在限制。可以從詳細的說明書和從權利要求書容易地看到本發明的其它特徵和優點。為了進一步定義本發明,提供以下術語和定義。在本文中所使用的「抗體」是指完整的免疫球蛋白或其抗原結合片段。本發明的抗體可以是任何同種型(isotype)或類(例如M、D、G、E和A)或任何亞類(例如G1-4、A1-2),並且可以具有或者kappa(κ)或者lambda(λ)輕鏈。在本文中所使用的術語「有效量」是指在劑量上和針對必需的時間段有效實現所需結果的量。所需結果可以是例如體外或體內減少或抑制新生血管形成或血管生成。有效量不需要是完全去除新生血管形成或血管生成。在本文中所使用的「治療有效量」是指在劑量上和針對必需的時間段有效實現所需治療結果的量。治療結果可以是例如症狀減輕、在放射成像中腫瘤尺寸的消退或穩定化、延長的存活期、改善的運動性等。治療結果不需要是「治癒」。在本文中所使用的「預防有效量」是指在劑量上和針對必需的時間段有效實現所需預防結果的量。典型地,由於預防劑量是在疾病的很早期之前或在疾病的早期被用於受試者,所述預防有效量將小於所述治療有效量。術語「多核苷酸」或「核苷酸」涵蓋單數核酸以及複數核酸,並且指分離的核酸分子或構建體,例如信使RNA(mRNA)或質粒DNA(pDNA)。在某些實施方案中,多核苷酸包含常規磷酸二酯鍵或非常規的鍵(例如醯胺鍵,如發現於肽核酸(PNA)中的鍵)。在本文中所使用的「多核苷酸」、「核苷酸」、「核酸」可以互換使用並含有全長cDNA序列的核苷酸序列,包括非翻譯5'和3'序列、編碼序列以及所述核酸序列的片段、表位、結構域和變體。所述多核苷酸可以由任意多核糖核苷酸或多脫氧核糖核苷酸組成,其可以是未修飾的RNA或DNA或者修飾的RNA或DNA。例如,多核苷酸可以由單鏈和雙鏈DNA、單鏈和雙鏈區域混合的DNA、單鏈和雙鏈RNA、單鏈和雙鏈區域混合的RNA、包含可以是單鏈或更典型地雙鏈或單鏈和雙鏈區域混合的DNA和RNA的雜合分子組成。此外,所述多核苷酸可以由包含RNA或DNA或RNA和DNA二者的三鏈區域組成。多核苷酸還可以含有一個或更多個修飾的鹼基或因為穩定性或因為其它原因被修飾的DNA或RNA骨架。「修飾的」鹼基包括例如三苯甲基化鹼基和稀有鹼基(如肌苷)。可以對DNA和RNA進行各種修飾;因此,「多核苷酸」包括化學、酶法或代謝修飾的形式。在本發明中,多肽可以由通過肽鍵或修飾的肽鍵(即肽電子等排物)互相結合的胺基酸組成,並可以含有除20種基因編碼的胺基酸之外的胺基酸(例如非天然存在的胺基酸)。本發明的多肽可以或者通過天然的過程(如翻譯後加工)或者通過本領域公知的化學修飾技術進行修飾。這樣的修飾被充分描述於基礎教科書和更詳細的專著以及大量的研究文獻中。修飾可以在多肽的任何地方發生,包括肽骨架、胺基酸側鏈和氨基或羧基末端。應當理解的是,相同類型的修飾可以相同或不同的程度存在於給定多肽的幾個位點。而且,給定多肽可以含有許多類型的修飾。多肽可以是分支的,例如作為泛素化的結果,並且它們可以是環狀的,有或者沒有分支。環狀的、分支的和分支環狀多肽可以由翻譯後天然過程導致或者可以由合成方法進行製備。修飾包括乙醯化、醯化、ADP-核糖基化、醯胺化、黃素的共價連接、血紅素部分的共價連接、核苷酸或核苷酸衍生物的共價連接、脂質或脂質衍生物的共價連接、磷脂醯肌醇的共價連接、交聯、環化、二硫鍵形成、脫甲基化、共價交聯的形成、半胱氨酸的形成、焦穀氨酸的的形成、甲醯化、γ-羧化、糖基化、GPI錨的形成、羥基化、碘化、甲基化、豆蔻醯化、氧化、聚乙二醇化、蛋白質水解加工、磷酸化、異戊二烯化、外消旋化、硒化、硫酸化、轉移RNA介導胺基酸添加到蛋白質(如精氨醯化)和泛素化。(參見,例如Proteins-StructureAndMolecularProperties,2ndEd.,T.E.Creighton,W.H.FreemanandCompany,NewYork(1993);PosttranslationalCovalentModificationofProteins,B.C.Johnson,Ed.,AcademicPress,NewYork,pgs.1-12(1983);Seifteretal.,MethEnzymol182:626-646(1990);Rattanetal.,AnnNYAcadSci663:48-62(1992)。)當涉及本發明的任意多肽或多核苷酸時,術語「片段」、「變體」、「衍生物」和「類似物」包括保留至少一些活性(即作為所述多肽或多核苷酸的任意天然存在的功能發揮作用的能力)的任意多肽或多核苷酸。例如腫瘤壞死因子受體1(TNFR1)的「片段」、「變體」、「衍生物」和「類似物」具有天然存在的全長TNFR1的一些活性,例如,結合TNFR1配體(即TNF-α或淋巴毒素)的能力。在另一個實例中,FAS多肽的「片段」、「變體」、「衍生物」和「類似物」具有天然存在的全長FAS多肽的一些活性,例如,誘導細胞凋亡的能力。在其它實例中,內皮細胞特異性啟動子的「片段」、「變體」、「衍生物」和「類似物」能夠誘導被可操作地連接至所述啟動子的基因的內皮細胞特異性表達。TNFR1、FAS多肽和內皮細胞特異性啟動子的各種片段、變體、類似物或衍生物的另外的非限制性的實例描述如下。在本發明中,「多肽片段」或「蛋白質片段」是指多肽的短胺基酸序列。蛋白質或多肽片段可以是「獨立式的」,或包含於更大的多肽內,其中所述片段形成區域的一部分。本發明的多肽片段的代表性實例包括例如包含大約5個胺基酸、大約10個胺基酸、大約15個胺基酸、大約20個胺基酸、大約30個胺基酸、大約40個胺基酸、大約50個胺基酸、大約60個胺基酸、大約70個胺基酸、大約80個胺基酸、大約90個胺基酸和大約100個胺基酸的片段。「保守性胺基酸置換」是這樣的,其中所述胺基酸殘基被替換為具有類似側鏈的胺基酸殘基。在本領域已經定義了具有類似側鏈的胺基酸殘基的家族,包括鹼性側鏈(例如賴氨酸、精氨酸、組氨酸)、酸性側鏈(例如天冬氨酸、穀氨酸)、不帶電荷的極性側鏈(例如甘氨酸、天冬醯胺、穀氨醯胺、絲氨酸、蘇氨酸、酪氨酸、半胱氨酸)、非極性側鏈(例如丙氨酸、纈氨酸、亮氨酸、異亮氨酸、脯氨酸、苯丙氨酸、甲硫氨酸、色氨酸)、β-分支側鏈(例如蘇氨酸、纈氨酸、異亮氨酸)和芳香族側鏈(例如酪氨酸、苯丙氨酸、色氨酸、組氨酸)。因此,如果多肽中的胺基酸被替換成來自相同側鏈家族的另一胺基酸,則該置換被認為是保守的。在另一個實施方案中,一串胺基酸可以被保守替換成側鏈家族成員的順序和/或組成不同的結構類似的一串胺基酸。兩個多核苷酸或多肽序列之間的術語「序列同一性百分比」是指,考慮到針對這兩個序列的最佳比對必須被引入的添加或缺失(即缺口),在比較窗口上由所述序列共有的相同匹配位置的數量。匹配位置是靶序列和參考序列中均表現相同的核苷酸或胺基酸的任意位置。不計算在靶序列中表現的缺口,因為缺口不是核苷酸或胺基酸。同樣,不計算在參考序列中表現的缺口,因為計算的是靶序列的核苷酸或胺基酸,而不是來自參考序列的核苷酸或胺基酸。通過以下方法計算序列同一性的百分比:確定在兩個序列中均存在相同的胺基酸殘基或核酸鹼基的位置的數量以得到匹配位置的數量,將匹配位置的數量除以比較窗口中位置的總數並將結果乘以100,得到序列同一性的百分比。兩個序列之間的序列比較和序列同一性百分比的確定可以使用對於在線使用和下載均容易獲得的軟體來完成。合適的軟體程序可以從各種來源獲得,並且可用於蛋白質和核苷酸序列的比對。適於確定序列同一性百分比的一個程序是bl2seq,該程序為可從美國政府的國家生物技術信息中心BLAST網站(blast.ncbi.nlm.nih.gov)獲得的BLAST程序套件的部分。Bl2seq使用或者BLASTN或者BLASTP算法進行兩個序列之間的比較。BLASTN是用於比較核酸序列,而BLASTP是用於比較胺基酸序列。其它合適的程序有例如Needle、Stretcher、Water或Matcher,其為EMBOSS生物信息程序套件的部分,並且也可從歐洲生物信息研究所(EBI)在www.ebi.ac.uk/Tools/psa獲得。與多核苷酸或多肽參考序列比對的單個多核苷酸或多肽靶序列內的不同區域可以各自具有它們自己的序列同一性百分比。應注意的是,序列同一性百分比的值捨入至最接近的十分之一。例如,80.11、80.12、80.13和80.14向下捨入至80.1,而80.15、80.16、80.17、80.18和80.19向上捨入至80.2。還應注意的是,長度的值將始終是整數。本領域技術人員將理解的是,用於計算序列同一性百分比的序列比對的產生並不僅僅局限於完全由原始序列數據所驅動的二元序列-序列比較。序列比對可以源自多重序列比對。適於產生多個序列比對的一個程序是ClustalW2,其可從www.clustal.org獲得。另一個合適的程序是MUSCLE,其可從www.drive5.com/muscle/獲得。ClustalW2和MUSCLE備選地可從例如EBI獲得。還將理解的是,可以通過將序列數據與異質來源(如結構數據(例如晶體學蛋白質結構)、功能數據(例如突變的位置)或系統發生數據)整合來產生序列比對。整合異質數據以產生多重序列比對的合適的程序是T-Coffee,其可從www.tcoffee.org獲得,並且備選地可從例如EBI獲得。還將理解的是,用於計算序列同一性百分比的最終比對可以被或者自動或者手工輔助。在本文中所使用的術語「連接的」、「融合的(fused)」、「融合/融合體(fusion)」、「嵌合的」和「嵌合」可以互換使用。這些術語是指兩個或更多個的元件或成分以任何方式(包括化學綴合或重組方式)連接在一起。「框內融合」是指兩個或更多個開放閱讀框(ORF)以保持原始ORF的正確閱讀框的方式連接而形成連續的更長ORF。因此,所得到的重組融合體或嵌合蛋白質是含有對應於由原始ORF編碼的多肽的兩個或更多個區段的單個蛋白質(所述區段在自然界中正常情況下是不會這樣連接的)。儘管由此所製得的閱讀框在整個融合區段上是連續的,但是這些區段可以由例如框內接頭序列在物理上或空間上分隔開。術語「異源的」和「異源部分」是指源自與其要比較的實體不同的實體的多核苷酸、多肽或其它部分。例如,異源多肽可以是合成的或源自不同物種、個體的不同細胞類型或不同個體的相同或不同類型的細胞。在一個方面,異源部分可以是被融合至另一個多肽以產生融合多肽或蛋白質的多肽。在另一個方面,異源部分可以是被綴合至多肽或蛋白質的非多肽(如PEG)。在多肽的上下文中,「線性序列」或「序列」是多肽中以從氨基到羧基末端方向的胺基酸的順序,其中所述序列中彼此相鄰的殘基在所述多肽的一級結構中是連續的。在本文中所使用的術語「表達」是指基因通過其產生生化產品(例如RNA或多肽)的過程。所述過程包括細胞內所述基因的功能性存在的任何表現,包括但不限於基因敲低以及瞬時表達和穩定表達。它包括但不限於基因轉錄成信使RNA(mRNA)、轉運RNA(tRNA)、小髮夾RNA(shRNA)、小幹擾RNA(siRNA)或任何其它RNA產物和這樣的mRNA翻譯成一個或多個多肽。如果最終所需產物是生化產品,表達包括創造該生化產品和任何前體。二、包含FAS嵌合基因和內皮細胞特異性啟動子的核酸構建體本發明涉及通過抑制、降低或減少腫瘤中血管生成或新生血管形成來減少或降低女性婦科癌症中腫瘤的尺寸的方法,該方法包括施用表達FAS嵌合蛋白的核酸構建體。編碼FAS嵌合蛋白(或基因產物)的基因在本發明中可被連接至指導所述FAS嵌合基因產物在內皮細胞中表達的內皮細胞特異性啟動子。這樣的細胞毒性基因產物的表達在不期望有過量的新生血管形成或血管生長的情況下(例如在腫瘤中)是有用的。本發明還提供包含被可操作地連接至內皮細胞特異性啟動子的FAS嵌合基因的核酸構建體的均質群體。A.FAS嵌合由本發明的核酸構建體表達的FAS嵌合蛋白包含至少兩個「死亡受體」多肽,所述多肽的每一個源自不同的蛋白質。FAS嵌合蛋白的第一多肽包含腫瘤壞死因子受體1(TNFR1)的配體結合結構域。FAS嵌合蛋白的第二多肽包含FAS多肽的效應子結構域。TNFR1的配體結合結構域可以是結合TNFR1配體的任何結構域。在一個實施方案中,TNFR1配體是TNF-α。在另一個實施方案中,TNFR1配體是淋巴毒素-α。TNFR1的配體結合結構域可以是TNFR1的胞外結構域或其任意片段、變體、衍生物或類似物。TNFR1配體結合結構域的非限制性的實例描述如下。可用於本發明的FAS多肽的效應子結構域包含形成死亡誘導性信號傳導複合物(death-inducingsignalingcomplex,DISC)的任何FAS結構域,從而誘導細胞凋亡。在一個實施方案中,FAS多肽的效應子結構域包含胞內結構域、跨膜結構域或二者均包含。FAS多肽效應子結構域的非限制性的實例描述如下。TNFR1和FAS多肽可以通過肽鍵或通過接頭進行連接。將TNFR1配體結合結構域與FAS效應子結構域連接的接頭可以是多肽接頭或非肽接頭。例如,用於FAS嵌合蛋白的接頭可以包含一個或更多個甘氨酸、絲氨酸、亮氨酸或其任意組合。在一個實施方案中,可用於本發明的接頭包含Ser-Leu。在另一個實施方案中,可用於本發明的接頭包含(GGGS)n,(Deniseetal.J.Biol.Chem.277:35035-35043(2002)),其中n可以是0、1、2、3、4、5、6、7、8、9或10或更大(SEQIDNO:27)。1.腫瘤壞死因子受體1全長的人TNFR1多肽長455個胺基酸並且也被稱為TNF-R1、I型腫瘤壞死因子受體(TNFRI)、TNFR-I、TNFRSF1A、TNFAR、p55、P60或CD120a。已知天然存在的人TNFR1多肽結合TNF-α或同源三聚體淋巴毒素-α。TNF-α與胞外結構域的結合導致TNFR1的同源三聚化,然後其與1型腫瘤壞死因子受體相關死亡結構域蛋白(TRADD)的死亡結構域發生特異性相互作用。各種TRADD相互作用蛋白(如TNF受體相關因子(TRAFS)、受體相互作用性絲氨酸/蘇氨酸蛋白激酶1(RIPK1)和具有死亡結構域的Fas相關蛋白(FADD))通過它們與TRADD的締合被募集至複合物。該複合物活化至少兩個不同的信號級聯,細胞凋亡和NF-κ-B信號傳導。作為人TNFR1多肽序列報導的455個胺基酸的多肽序列在UniProtKB資料庫中具有識別號P19438-1。在本文中該人TNFR1多肽序列被命名為同工型(isoform)A和SEQIDNO:2。SEQIDNO:1是編碼SEQIDNO:2的核苷酸序列。108aa的多肽序列被報導為人TNFR1多肽序列的同工型並且在UniProtKB資料庫中具有識別號P19438-2。該108aa的多肽對應於同工型A(SEQIDNO:2)的第1至108位胺基酸並且在本文中被命名為同工型B。具有232aa的人TNFR1多肽的另一個變體被報導為在UniProtKB資料庫中具有識別號P19438-3。該232aa的多肽對應於同工型A(SEQIDNO:2)的第1至232位胺基酸並且在本文中被命名為同工型C。人TNFR1的另外的天然變體包括但不限於包含選自由以下突變組成的組的一個或更多個突變的同工型A、B和C的TNFR1多肽:H51Q、C59R、C59S、C62G、C62Y、P75L、T79M、C81F、C99S、S115G、C117R、C117Y、R121P、R121Q、P305T及其任意組合。其它已知TNFR1變體包括包含L13LILPQ、K255E、S286G、R394L、412:缺失、GPAA443-446APP或其任意組合的同工型A、B和C的TNFR1多肽。表1顯示了人野生型TNFR1胺基酸序列和編碼所述野生型TNFR1的核苷酸序列。表1.TNFR1序列本發明還報導了小鼠TNFR1多肽序列及其變體。454aa的小鼠TNFR1多肽在UniProtKB資料庫中具有識別號P25118。在其它動物中已知的TNFR1多肽包括但不限於大鼠(例如UniProtKB資料庫中的P22934)、牛(例如UniProtKB資料庫中的O19131)、豬(例如UniProtKB資料庫中的P50555)或馬(例如UniProtKB資料庫中的D1MH71)。全長的TNFR1能夠被裂解成兩條鏈,(1)TNF受體超家族成員1A、膜形式(即對應於全長TNFR1的第22至455位胺基酸)和(2)TNF結合蛋白1(TBPI)(即對應於全長TNFR1的第41至291位胺基酸)。全長的人TNFR1多肽由信號序列(SEQIDNO:2的第1至21位胺基酸)、胞外結構域(SEQIDNO:2的第22至211位胺基酸)、跨膜結構域(SEQIDNO:2的第212至234位胺基酸)和胞質結構域(SEQIDNO:2的第235至455位胺基酸)組成。TNFR1的胞外結構域包含四個半胱氨酸重複區域,TNFR-Cys1(對應於SEQIDNO:2的第43至82位胺基酸)、TNFR-Cys2(對應於SEQIDNO:2的第83至125位胺基酸)、TNFR-Cys3(對應於SEQIDNO:2的第126至166位胺基酸)和TNFR-Cys4(對應於SEQIDNO:2的第167至196位胺基酸)。如本領域技術人員將理解的,以上所列的結構域的開始和結束的殘基可以改變,這取決於所使用的計算機建模程序或用於確定該結構域的方法。因此,TNFR1的各種功能結構域可以與上述定義的那些不同。在一個實施方案中,可用於FAS嵌合蛋白的TNFR1的配體結合結構域包含、TNFR1的胞外結構域或其任何片段、變體、衍生物或類似物基本上由其組成或者由其組成,其中所述TNFR1的胞外結構域或其任何片段、變體、衍生物或類似物結合TNF-α。在另一個實施方案中,TNFR1的配體結合結構域包含TNFR-Cys1;TNFR-Cys2;TNFR-Cys3;TNFR-Cys4;TNFR-Cys1和TNFR-Cys2;TNFR-Cys1和TNFR-Cys3;TNFR-Cys1和TNFR-Cys4;TNFR-Cys2和TNFR-Cys3;TNFR-Cys2和TNFR-Cys4;TNFR-Cys3和TNFR-Cys4;TNFR-Cys1,TNFR-Cys2和TNFR-Cys3;TNFR-Cys1,TNFR-Cys2和TNFR-Cys4;TNFR-Cys2,TNFR-Cys3和TNFR-Cys4;或TNFR-Cys1,TNFR-Cys2,TNFR-Cys3和TNFR-Cys4。在其它實施方案中,FAS嵌合蛋白中的TNFR1配體結合結構域包含TNF結合蛋白I。在又一些其它實施方案中,FAS嵌合蛋白的TNFR1配體結合結構域包含與SEQIDNO:2的第22至190位胺基酸、第22至191位胺基酸、第22至192位胺基酸、第22至193位胺基酸、第22至194位胺基酸、第22至195位胺基酸、第22至196位胺基酸、第22至197位胺基酸、第22至198位胺基酸、第22至199位胺基酸、第22至200位胺基酸、第22至201位胺基酸、第22至202位胺基酸、第22至203位胺基酸、第22至204位胺基酸、第22至205位胺基酸、第22至206位胺基酸、第22至207位胺基酸、第22至208位胺基酸、第22至209位胺基酸、第22至210位胺基酸或第22至211位胺基酸至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列、基本上由其組成或者由其組成,其中所述配體結合結構域結合TNFR1配體,例如TNF-α。在其它實施方案中,所述TNFR1的配體結合結構域還包含信號肽。合適的信號肽的一個實例是TNFR1的信號肽,例如SEQIDNO:2的第1至21位胺基酸。在又一些其它實施方案中,FAS嵌合基因產物的配體結合結構域包含與SEQIDNO:2的第1至190位胺基酸、第1至191位胺基酸、第1至192位胺基酸、第1至193位胺基酸、第1至194位胺基酸、第1至195位胺基酸、第1至196位胺基酸、第1至197位胺基酸、第1至198位胺基酸、第1至199位胺基酸、第1至200位胺基酸、第1至201位胺基酸、第1至202位胺基酸、第1至203位胺基酸、第1至204位胺基酸、第1至205位胺基酸、第1至206位胺基酸、第1至207位胺基酸、第1至208位胺基酸、第1至209位胺基酸、第1至210位胺基酸或第1至211位胺基酸至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列、基本上由其組成或者由其組成,其中所述配體結合結構域結合TNFR1配體,例如TNF-α。在一個具體的實施方案中,FAS嵌合蛋白的TNFR1配體結合結構域包含與SEQIDNO:4至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列、基本上由其組成或者由其組成,其中所述配體結合結構域結合TNFR1配體,例如TNF-α。在又一些其它實施方案中,TNFR1的配體結合結構域由與SEQIDNO:3至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列編碼。在又一些其它實施方案中,FAS嵌合蛋白的TNFR1配體結合結構域包含與SEQIDNO:2的第22至108位胺基酸(TNFR1同工型B)、SEQIDNO:2的第22至232位胺基酸(TNFR1同工型C)或SEQIDNO:2的第44至291位胺基酸(TBP1)至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列、基本上由其組成或者由其組成,其中所述配體結合結構域結合TNFR1配體,例如TNF-α。2.FAS多肽全長的人FAS多肽長335個胺基酸,並且也被稱為腫瘤壞死因子受體超家族成員6、Apo-1抗原、細胞凋亡介導的表面抗原FAS、FASLG受體或CD95。天然存在的FAS多肽是針對TNFSF6/FASLG的受體。當FAS多肽結合FAS配體(FasL)時,FAS和FasL之間的相互作用導致形成死亡誘導性信號傳導複合物(DISC),其含有FADD、胱天蛋白酶-8和胱天蛋白酶-10。在一些細胞類型(I型)中,加工的胱天蛋白酶-8直接活化胱天蛋白酶家族的其它成員,並觸發細胞凋亡的執行。在其它細胞類型(II型)中,FAS-DISC啟動反饋環路,其螺旋形提高來自線粒體的促凋亡因子的釋放和胱天蛋白酶-8的放大的活化。FAS介導的細胞凋亡可能在外周耐受性的誘導中、在成熟細胞的抗原刺激的自殺中或者二者中發揮作用。作為人FAS多肽序列報導的335aa的多肽序列在UniProtKB資料庫中具有識別號P25445-1。該人FAS多肽序列被命名為SEQIDNO:6。SEQIDNO:5是編碼SEQIDNO:6的核苷酸序列。編碼FAS多肽的核苷酸序列也被稱為APT1、FAS1或TNFRSF6。全長FAS多肽含有信號肽(對應於SEQIDNO:6的第1至25位胺基酸)、胞外結構域(對應於SEQIDNO:6的第26至173位胺基酸)、跨膜結構域(對應於SEQIDNO:6的第174至190位胺基酸)和胞內(或胞質)結構域(對應於SEQIDNO:6的第191至335位胺基酸)。胞內結構域含有死亡結構域(例如對應於SEQIDNO:6的第230至314位胺基酸)。如本領域技術人員將理解的,以上所列的結構域的開始和結束的殘基可以因所使用的計算機建模程序或用於確定該結構域的方法而有所變化。因此,FAS的各種功能結構域可以由上述定義那些而變化。表2顯示了野生型人FAS胺基酸序列和編碼所述FAS的核苷酸序列。表2.FAS序列本發明還報導了小鼠FAS的多肽序列及其變體。327aa的小鼠FAS多肽在UniProtKB資料庫中具有識別號P25446。在其它動物中已知的FAS多肽包括但不限於舊大陸猴(例如UniProtKB資料庫中的Q9BDN4)、恆河猴(例如UniProtKB資料庫中的Q9BDP2)、大鼠(例如UniProtKB資料庫中的Q63199)或牛(例如UniProtKB資料庫中的P51867)。基於FAS多肽中的序列變化,本領域技術人員能夠鑑定FAS多肽的效應子結構域中的序列變化。例如,FAS效應子結構域的天然變體可以包括C178R、L180F、P183L、I184V、T198I、Y232C、T241K、T241P、V249L、R250P、R250Q、G253D、G253S、N255D、A257D、I259R、D260G、D260V、D260Y、I262S、N264K、T270I、T270K、E272G、E272K、L278F、K299N、T305I、I310S或其任意組合的一個或更多個置換或突變。在一個實施方案中,可用於本發明的FAS多肽的效應子結構域包含FAS多肽的死亡結構域。在另一個實施方案中,FAS多肽的效應子結構域包含與SEQIDNO:6的第230至314位胺基酸至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列、基本上由其組成或者由其組成。在其它實施方案中,FAS多肽的效應子結構域包含FAS多肽的胞內結構域。在又一些其它實施方案中,FAS多肽的效應子結構域包含與SEQIDNO:6的第185至335位胺基酸、第186至335位胺基酸、第187至335位胺基酸、第188至335位胺基酸、第189至335位胺基酸、第190至335位胺基酸、第191至335位胺基酸、第192至335位胺基酸、第193至335位胺基酸、第194至335位胺基酸、第195至335位胺基酸、第196至335位胺基酸、第197至335位胺基酸、第198至335位胺基酸、第199至335位胺基酸至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列。在又一些其它實施方案中,FAS多肽的效應子結構域還包含FAS多肽的跨膜結構域。在又一些其它實施方案中,FAS多肽的效應子結構域包含與SEQIDNO:6的第174至335位胺基酸至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列。在一些實施方案中,FAS多肽的效應子結構域還包含來自FAS胞外結構域的C末端部分的大約十個、大約九個、大約八個、大約七個、大約六個、大約五個、大約四個、大約三個、大約兩個或大約一個胺基酸。在某些實施方案中,FAS多肽的效應子結構域包含與SEQIDNO:6的第179至335位胺基酸、第178至335位胺基酸、第177至335位胺基酸、第176至335位胺基酸、第175至335位胺基酸、第174至335位胺基酸、第173至335位胺基酸、第172至335位胺基酸、第171至335位胺基酸、第170至335位胺基酸、第169至335位胺基酸、第168至335位胺基酸、第167至335位胺基酸、第166至335位胺基酸、第165至335位胺基酸、第164至335位胺基酸或第163至335位胺基酸至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列,其中所述效應子結構域形成死亡誘導性信號傳導複合物(DISC)、活化胱天蛋白酶8或誘導細胞凋亡。在一個實施方案中,FAS多肽的效應子結構域包含與SEQIDNO:8至少70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列、基本上由其組成或者由其組成,其中所述效應子結構域形成死亡誘導性信號傳導複合物(DISC)、活化胱天蛋白酶8或誘導細胞凋亡。在其它實施方案中,FAS多肽的效應子結構域由與SEQIDNO:7至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列編碼。在一個實施方案中,用於本發明的FAS嵌合基因產物包含與SEQIDNO:10至少70%、80%、90%、95%、96%、97%、98%、99%或100%相同的胺基酸序列、基本上由其組成或者由其組成,其中所述FAS嵌合基因產物誘導細胞凋亡。在另一個實施方案中,所述FAS嵌合基因產物由與SEQIDNO:9至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列編碼,其中所述FAS嵌合基因產物誘導細胞凋亡。B.內皮細胞特異性啟動子包含FAS嵌合基因的核酸構建體還包含一個或更多個可用於調控可操作地連接的FAS嵌合基因的表達的表達控制元件。所述表達控制元件包括但不限於啟動子、分泌信號和其它調控元件。可用於本發明的核酸構建體利用內皮細胞特異性啟動子指導FAS嵌合蛋白在內皮細胞中表達,從而誘導所述內皮細胞的細胞凋亡。為了本發明的目的,內皮細胞特異性啟動子可含有一個或更多個順式調控元件,其與不具有所述順式調控元件的啟動子相比改善啟動子的內皮細胞特異性。在一個實例中,所述順式調控元件包含增強子。在另一個方面,所述順式調控元件包含缺氧反應元件。在其它實例中,所述順式調控元件既包含增強子又包含缺氧反應元件。在一個實施方案中,可用於本發明的增強子包含與SEQIDNO:11或SEQIDNO:12(SEQIDNO:11的互補序列)至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列,其中,與不具有所述增強子的啟動子相比,所述增強子改善啟動子的內皮細胞特異性。所述增強子還可以包含與SEQIDNO:13或SEQIDNO:14(SEQIDNO:13的互補序列)至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的另外的核苷酸序列。在另一個實施方案中,用於本發明的增強子包含與SEQIDNO:13或SEQIDNO:14(SEQIDNO:13的互補序列)至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列,其中,與不具有所述增強子的啟動子相比,所述增強子改善啟動子的內皮細胞特異性。所述增強子還可以包含與SEQIDNO:11或SEQIDNO:12(SEQIDNO:11的互補序列)至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的另外的核苷酸序列。在其它實施方案中,用於本發明的增強子包含與SEQIDNO:15或SEQIDNO:16(SEQIDNO:15互補序列)至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列,其中,與不具有所述增強子的啟動子相比,所述增強子改善啟動子的內皮細胞特異性。在又一些其它實施方案中,用於所述核酸構建體的增強子包含SEQIDNO:15或SEQIDNO:16或其任何片段、變體、衍生物或類似物,其中,與不具有所述增強子的啟動子相比,所述片段、變體、衍生物或類似物改善啟動子的內皮細胞特異性。在一些實施方案中,用於本發明的增強子包含與SEQIDNO:22或SEQIDNO:23至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列,其中,與不具有所述增強子的啟動子相比,所述增強子改善啟動子的內皮細胞特異性。在又一些其它實施方案中,用於所述核酸構建體的增強子包含SEQIDNO:22或SEQIDNO:23或其任何片段、變體、衍生物或類似物,其中,與不具有所述增強子的啟動子相比,所述片段、變體、衍生物或類似物改善啟動子的內皮細胞特異性。在其它實施方案中,用於本發明的增強子包含與SEQIDNO:24或SEQIDNO:25至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%相同的核苷酸序列,其中,與不具有所述增強子的啟動子相比,所述增強子改善啟動子的內皮細胞特異性。在又一些其它實施方案中,用於所述核酸構建體的增強子包含SEQIDNO:24或SEQIDNO:25或其任何片段、變體、衍生物或類似物,其中,與不具有所述增強子的啟動子相比,所述片段、變體、衍生物或類似物改善啟動子的內皮細胞特異性。表3顯示了可用於本發明的各種增強子序列。表3.內皮細胞特異性增強子元件和啟動子用於本發明的增強子可以被連接至啟動子上遊或啟動子下遊或插入啟動子中的兩個核苷酸之間。用於本發明的內皮細胞特異性啟動子可以利用現有技術已知的任何啟動子。例如,能夠用於本發明的合適的啟動子包括內皮特異性啟動子:前內皮素原-1(preproendothelin-1,PPE-1啟動子)(US2010/0282634,公開於2010年11月11日;和WO2011/083464,公開於2011年7月14日);PPE-1-3X啟動子(美國專利號7,579,327,美國專利號8,071,740,US8,039,261,US2010/0282634,US2007/0286845,WO2011/083464和WO2011/083466);TIE-1(S79347,S79346)和TIE-2(U53603)啟動子[SatoTN,ProcNatlAcadSciUSA1993Oct15;90(20):9355-8],內皮糖蛋白(Endoglin)啟動子[Y11653;RiusC,Blood1998Dec15;92(12):4677-90],vonWillerbrand因子[AF152417;CollinsCJProcNatlAcadSciUSA1987Jul;84(13):4393-7],KDR/flk-1啟動子[X89777,X89776;RonickeV,CircRes1996Aug;79(2):277-85],FLT-1啟動子[D64016AJ224863;MorishitaK,:JBiolChem1995Nov17;270(46):27948-53],Egr-1啟動子[AJ245926;SukhatmeVP,OncogeneRes1987Sep-Oct;l(4):343-55],E-選擇素啟動子[Y12462;CollinsTJBiolChem1991Feb5;266(4):2466-73],內皮粘附分子啟動子:ICAM-1[X84737;HorleyKJEMBOJ1989Oct;8(10):2889-96]、VCAM-1[M92431;IademarcoMF,JBiolChem1992Aug15;267(23):16323-9]、PECAM-1[AJ313330X96849;CD31,NewmanPJ,Science1990Mar9;247(4947):1219-22],血管平滑肌特異性元件:CArG盒X53154和主動脈羧肽酶樣蛋白(ACLP)啟動子[AF332596;LayneMD,CircRes.2002;90:728-736]和主動脈優先表達基因-1[Yen-HsuChenJ.Biol.Chem,Vol.276,Issue50,47658-47663,December14,2001],其全部內容通過引用整體併入本文。在一個實施方案中,被連接至內皮細胞特異性增強子的啟動子包含SEQIDNO:17的至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%的核苷酸序列,其中所述被連接至增強子的啟動子誘導被可操作地連接至所述啟動子的基因的內皮細胞特異性。在另一個實施方案中,被連接至內皮細胞特異性增強子的啟動子包含野生型PPE-1啟動子的片段、變體、衍生物或類似物,其中其所述片段、變體、衍生物或類似物誘導被可操作地連接至所述啟動子的基因的內皮細胞特異性。在一個實例中,該內皮細胞特異性增強子能夠被插入對應於SEQIDNO:17的第442位和第449位核苷酸殘基之間。在進一步的實施方案中,內皮細胞特異性啟動子包含缺氧反應元件。缺氧反應元件(HRE)位於內皮素(endothelin)-1啟動子的反義鏈上。該元件是缺氧誘導性因子-1的結合位點,該位點是缺氧對(人、大鼠和小鼠基因的)內皮素-1啟動子的正向調控所必需的。缺氧是強效信號,其誘導幾種基因的表達,包括促紅細胞生成素(Epo)、VEGF和各種糖酵解酶。在所有基因中響應缺氧條件的核心序列(8個鹼基對)是保守的,側翼區與其它基因不同。ET-I缺氧反應元件位於GATA-2和AP-1結合位點之間。在一個實例中,缺氧反應元件包含SEQIDNO:26、其片段、變體、衍生物或類似物。在其它實施方案中,可用於本發明的內皮細胞特異性啟動子包含SEQIDNO:18的至少60%、70%、80%、90%、95%、96%、97%、98%、99%或100%的核苷酸序列、基本上由其組成或者由其組成,其中被連接至增強子的啟動子誘導被可操作地連接至所述啟動子的基因的內皮細胞特異性。在另一個實施方案中,內皮細胞特異性啟動子包含SEQIDNO:18的片段、變體、衍生物或類似物,其中所述片段、變體、衍生物或類似物誘導被可操作地連接至所述啟動子的基因的內皮細胞特異性。內皮細胞特異性啟動子的另外的變化可以被發現於WO2011/083464、WO2011/083466和WO2012/052423,其通過引用整體併入本文。本發明還提供包含核苷酸序列SEQIDNO:17的新的啟動子序列。在一個實例中,所述啟動子還包含內皮細胞特異性增強子。在一個實例中,所述內皮細胞特異性增強子包含SEQIDNO:11,SEQIDNO:12,SEQIDNO:13,SEQIDNO:14,SEQIDNO:15,SEQIDNO:16,SEQIDNO:22,SEQIDNO:23,SEQIDNO:24,SEQIDNO:25,SEQIDNO:26或其任意片段、變體、衍生物或類似物,其中,與不具有所述增強子的啟動子相比,所述片段、變體、衍生物或類似物改善啟動子的內皮細胞特異性。在另一個實例中,所述啟動子包含SEQIDNO:18的核苷酸序列。本發明包括包含該新的啟動子和異源核苷酸序列的核酸構建體。在一個實施方案中,所述異源核酸序列包含編碼本文所述的FAS嵌合蛋白的核苷酸序列。在另一個實施方案中,所述異源核酸序列包含腺病毒序列。C.載體本發明還提供包含所述核酸構建體的載體,其包含被可操作地連接至內皮細胞特異性啟動子的FAS嵌合基因。為了本發明的目的,可以採用眾多載體系統。例如,可以用於本發明該方面的實施的各種病毒基因遞送系統包括但不限於腺病毒載體、α病毒載體、腸病毒載體、瘟病毒載體、慢病毒載體、杆狀病毒載體、皰疹病毒載體、EB病毒載體、乳多空病毒載體、痘病毒載體、痘苗病毒載體、腺相關病毒載體和單純皰疹病毒載體。在另一個實施方案中,包含被可操作地連接至內皮細胞特異性啟動子的FAS嵌合基因的載體是腺病毒。例如,所述腺病毒可以是人腺病毒A種(血清型12、18和31)、B種(血清型3、7、11、14、16、21、34、35、50和55)、C種(血清型1、2、5、6和57)、D種(血清型8、9、10、13、15、17、19、20、22-30、32、33、36-39、42-49、51、53、54和56)、E種(血清型4)、F種(血清型40和41)或G種(血清型52)中的任意一個或更多個。在一個具體的實施方案中,用於本發明的腺病毒是人腺病毒血清型5。在一些實施方案中,可用於基因療法的腺病毒是重組非複製型腺病毒,其不含有E1區域和E3區域。在某些實施方案中,用於本發明的腺病毒是條件複製型腺病毒,其不含有E3區域,但是含有E1區域。D.生物保藏根據布達佩斯條約並根據37C.F.R.§1.808由位於HealthProtectionAgencyCultureCollections,HealthProtectionAgency,MicrobiologyServices,PortonDown,Salisbury,SP40JG,UK的歐洲細胞培養物保藏中心(ECACC)進行生物保藏。一旦授予專利權,保藏材料的樣品將成為公眾可以獲得的樣品。因為所保藏的實施方案僅僅旨在舉例說明本發明,所以在本文中所述和所要求保護的發明並不限於所保藏的株的範圍。株ECACC登錄號藏日期VB-111130212012013年2月12日三、使用表達FAS嵌合蛋白的腺病毒的治療方法本發明的一個實施方案提供用於使用表達FAS嵌合蛋白的核酸構建體或包含所述核酸構建體的腺病毒的方法。在一個方面,表達FAS嵌合蛋白的核酸構建體或包含所述核酸構建體的腺病毒可用於在受試者中減少或降低腫瘤的尺寸或消除腫瘤,其中由所述核酸構建體編碼的FAS嵌合蛋白在所述受試者中減少或降低所述腫瘤的尺寸或減緩腫瘤生長速率或防止出現新的腫瘤轉移灶或消除所述腫瘤,並且其中所述腫瘤與女性婦科癌症或其轉移有關或源自所述女性婦科癌症或其轉移。可以基於放射診斷測試(如CT掃描)和/或腫瘤標誌物(如CA-125的血液水平)來評估這些效果。在另一個方面,表達FAS嵌合蛋白的核酸構建體或包含所述核酸構建體的腺病毒可用於抑制、降低或減少腫瘤中新生血管形成或血管生成,其中由所述核酸構建體編碼的FAS嵌合蛋白抑制、降低或減少腫瘤中新生血管形成或血管生成,並且其中所述腫瘤與女性婦科癌症或其轉移有關或源自所述女性婦科癌症或其轉移。在其它方面,表達FAS嵌合蛋白的核酸構建體或包含所述構建體的腺病毒能夠治療或預防受試者中與女性婦科癌症或其轉移有關或源自所述女性婦科癌症或其轉移的腫瘤,其中由所述核酸構建體編碼的FAS嵌合蛋白治療或預防所述受試者中的女性婦科癌症或其轉移。因此,在一個方面,本發明提供在受試者中減少或降低腫瘤或其轉移的尺寸、消除腫瘤或其轉移或者減緩腫瘤或其轉移的生長的方法,該方法包括給患者施用有效量的核酸構建體(其包含被可操作地連接至內皮細胞特異性啟動子的Fas嵌合蛋白)或包含所述核酸構建體的腺病毒,其中由所述核酸構建體編碼的Fas嵌合基因產物在受試者中減少或降低所述腫瘤或其轉移的尺寸或者消除所述腫瘤或其轉移,並且其中所述腫瘤或其轉移與女性婦科癌症有關或源自所述女性婦科癌症。在另一個方面,本發明提供抑制、降低或減少腫瘤或其轉移中新生血管形成或血管生成的方法,該方法包括給患有所述腫瘤或其轉移的受試者施用有效量的核酸構建體或包含所述核酸構建體的腺病毒,其包含被可操作地連接至內皮細胞特異性啟動子的Fas嵌合基因,其中由所述核酸構建體編碼的Fas嵌合基因產物抑制、減少或降低所述腫瘤或其轉移中新生血管形成或血管生成,並且其中所述腫瘤或其轉移與女性婦科癌症有關或源自所述女性婦科癌症。在其它方面,本發明提供治療或預防受試者中與女性婦科癌症有關或源自所述女性婦科癌症的腫瘤或其轉移的方法,該方法包括施用有效量的核酸構建體,其包含被可操作地連接至內皮細胞特異性啟動子的Fas嵌合基因,其中由所述核酸構建體編碼的Fas嵌合基因產物治療或預防患者中的女性婦科癌症。仍在一些其它實施方案中,所述女性婦科癌症或其轉移的腫瘤在所述施用之後被降低尺寸或被消除。在某些實施方案中,所述腫瘤或其轉移的尺寸被降低,從而與所述施用之前所述腫瘤的最大直徑(LD)相比,所述LD被降低至少大約10%、20%、30%、40%、50%、60%、70%、80%、90%或100%。在一些實施方案中,本發明包括穩定與女性婦科癌症有關的疾病或紊亂的方法。例如,本發明包括阻止或減緩與女性婦科癌症有關的腫瘤的進一步生長的方法。在進一步的實施方案中,本發明減少惡性腹膜液(例如腹水)的體積、減少受試者的疼痛、延長受試者的存活期或其任意組合。在其它實施方案中,本發明的腺病毒當被施用給受試者時延長該受試者的總體存活期。在進一步的實施方案中,本發明的腺病毒當被施用給受試者時延長該受試者的無進展存活期。在一個實施方案中,所述女性婦科癌症可以是米勒管癌、卵巢癌、腹膜癌、輸卵管癌、子宮乳頭狀漿液性癌、或其轉移、或其任意組合。在另一個實施方案中,所述女性婦科癌症可以是子宮頸癌、子宮內膜癌、妊娠滋養細胞疾病、子宮癌、外陰癌、或其轉移、或其任意組合。在其它實施方案中,所述女性婦科癌症包括從婦科組織(子宮、卵巢、輸卵管、子宮頸、卵細胞、支持細胞或其任意組合)所產生的任何癌性生長。在某些實施方案中,所述與女性婦科癌症有關或源自所述女性婦科癌症的腫瘤可以選自由以下組成的組:表面上皮間質瘤(腺癌)、乳頭狀漿液性囊腺癌、子宮內膜樣瘤、漿液性囊腺癌、乳頭狀瘤、粘液性囊腺癌、透明細胞卵巢瘤、粘液腺癌、囊腺癌、癌、性索間質瘤、生殖細胞瘤、畸胎瘤、無性細胞瘤、表皮樣瘤(鱗狀細胞癌)、布萊納瘤、或其轉移或其任意組合。用於本發明的目的的米勒管癌症包括惡性米勒管混合瘤,也被稱為惡性混合性中胚葉腫瘤、MMMT和癌肉瘤。MMMT是發現於子宮、卵巢、輸卵管以及身體其它部位的惡性腫瘤,其含有癌性(上皮組織)和肉瘤性(結締組織)成分。它被分成兩種類型,同源的(其中肉瘤性成分由發現於子宮中的組織(如子宮內膜、纖維和/或平滑肌組織)組成)和異源的類型(由不在子宮發現的組織(如軟骨、骨骼肌和/或骨)組成)。MMMT佔源自子宮體的所有腫瘤的百分之二至五之間,並且主要發現於平均年齡66歲的絕經後女性。卵巢癌包含從卵巢所產生的任何癌性生長。大多數(超過90%)卵巢癌被分類為「上皮」並且被認為從卵巢的表面產生。但是,輸卵管也可以是一些卵巢癌的來源。由於卵巢和輸卵管彼此密切相關,據認為這些輸卵管癌細胞能夠模仿卵巢癌。其它類型可以從卵細胞(生殖細胞瘤)或支持細胞產生。在一些實施方案匯總,卵巢癌是繼發性癌症,其為體內其它地方的原發癌症轉移的結果。大約7%的卵巢癌是由於轉移,而其餘的都是原發癌症。常見的原發癌症是乳腺癌和胃腸癌。腹膜癌(cancer)或癌(carcinoma)也被稱為:漿液性表面乳頭狀癌、原發腹膜癌、卵巢外漿液性癌、原發漿液性乳頭狀癌或沙樣癌。它在歷史上被分類至「不明原發癌」(CUP)之下。原發腹膜癌(PPC或PPCa)是腹膜或腹腔內膜細胞的癌。組織學和分子生物學特徵表明漿液性癌(其包括卵巢漿液性癌、子宮漿液性癌、輸卵管漿液性癌、宮頸漿液性癌和原發腹膜漿液性癌)事實上代表一個實體。原發輸卵管癌(PFTC)(往往只是輸卵管癌)是起源於輸卵管的惡性腫瘤。輸卵管癌被認為是女性中佔所有婦科癌症的1至2%的相對罕見的原發癌症。子宮漿液性癌(USC)(也被稱為子宮乳頭狀漿液性癌(UPSC)和子宮漿液性腺癌)是不常見的子宮內膜癌,其通常發生於絕經後女性。它通常在由絕經後出血提示的子宮內膜活檢中被診斷出。不同於更常見的低級子宮內膜樣子宮內膜腺癌,USC不會從子宮內膜增生發展並且不是激素敏感性的。它產生於子宮內膜萎縮的情況下並且被分類為II型子宮內膜癌。術語「受試者」或「個體」或「動物」或「患者」或「哺乳動物」是指任何受試者,特別是哺乳動物受試者,其患有或者預計發展與腹膜癌或女性婦科癌症有關或源自腹膜癌或女性婦科癌症的至少一種腫瘤。在一個實施方案中,所述受試者是人。在另一個實施方案中,所述受試者是癌症患者。在本發明的一個實施方案中,所述受試者是之前已經接受基於鉑的療法的患者。這樣的之前基於鉑的療法包括但不限於順鉑、卡鉑、奧沙利鉑、奈達鉑、沙鉑、吡鉑、四硝酸三鉑或aroplatin。基於鉑的抗腫瘤劑引起DNA的交聯如monadduct、鏈間交聯、鏈內交聯或DNA蛋白質交聯。多數情況下,它們作用於相鄰的鳥嘌呤的N-7位置,形成1,2鏈內交聯。所得到的交聯抑制癌細胞中的DNA修復和/或DNA合成。由於與烷基化抗腫瘤劑類似的效果,儘管它們不具有烷基基團,但是基於鉑的抗腫瘤劑有時被描述為「烷基化樣」。在某些實施方案中,之前的基於鉑的療法是使用順鉑(cisplatin)(其也被稱為順鉑(cisplatinum)或順-二胺二氯絡鉑(II)(CDDP)(由CadilaHealthcare根據FDA橙皮書銷售的商品名Cisplatin、商標名Platin))的療法。順鉑作為短期輸注以生理鹽水的形式進行靜脈內施用用於治療實體惡性腫瘤。它被用於治療各種類型的癌症,包括肉瘤、一些癌(例如小細胞肺癌和卵巢癌)、淋巴瘤和生殖細胞瘤。在其它實施方案中,所述受試者之前已經接受基於紫杉烷類的療法。紫杉烷類是由紫杉屬(紫杉)的植物產生的雙萜,並且被廣泛地用作化療劑。紫杉烷現在能夠人工合成。紫杉烷劑包括但不限於紫杉醇和多西他賽在一個方面,紫杉烷能夠被融合至或結合至異源部分。這樣的異源部分能夠改善紫杉烷製劑的溶解性或減少紫杉烷的毒性。例如,紫杉烷能夠被融合至或結合至白蛋白:白蛋白結合型紫杉醇(Abraxane,也被稱為nab-paclitaxel)是備選的製劑,其中紫杉醇結合至白蛋白納米顆粒。紫杉醇生產的合成方法導致多西他賽的開發。多西他賽與紫杉醇具有一套類似的臨床用途並且以的名稱進行銷售。在另一個方面,可用於本發明的紫杉烷包括但不限於榛植物的殼和葉中的紫杉醇、10-脫乙醯基巴苦亭III、巴苦亭III、紫杉醇C和7-表紫杉醇。在其它實施方案中,所述受試者之前已經接受高達三、高達二或高達一線的化療。前一線的化療可以是基於鉑的療法或基於紫杉烷的療法。在又一些其它實施方案中,所述受試者之前沒有接受超過3線的針對復發癌症的化療。在某些實施方案中,所述受試者是患有復發性鉑抗性癌症或對鉑無效(platinumrefractory)的疾病的患者。在一些實施方案中,所述受試者是患有復發性紫杉烷抗性癌症的患者。在一個方面,復發性鉑抗性癌症或復發性紫杉烷抗性癌症在鉑或紫杉烷治療期間,在完成或接受基於鉑的療法或基於紫杉烷的療法的大約一個月內、大約兩個月內、大約三個月、大約四個月、大約五個月、大約六個月、大約七個月、大約八個月、大約九個月、大約十個月、大約11個月或大約12個月內具有進展性腫瘤。在一個具體的實施方案中,所述復發性鉑抗性癌症或所述復發性紫杉烷抗性癌症在完成或接受基於鉑的療法或基於紫杉烷的療法的大約六個月內具有進展性腫瘤。所述復發性鉑抗性癌症或所述復發性紫杉烷抗性癌症可以由實體瘤響應評價標準(RECIST)、一個或更多個腫瘤標誌物(例如CA-125)的測定、體檢、重新評估或二探剖腹檢查和/或一種或更多種成像研究(例如X射線、CT或MRI)來確定。RECIST是一套出版的規則,其定義在治療期間什麼時候癌症患者改善(「響應」)、保持相同(「穩定」)或惡化(「進展」)。最初標準由包括歐洲癌症研究與治療組織(EORTC)、美國國家癌症研究所(NCI)和加拿大臨床試驗組的國家癌症研究所的國際合作組織於2000年2月出版。於2009年1月出版的RECIST1.1是最初標準的更新版。大多數評估癌症治療在實體瘤中的目標響應的臨床試驗正在使用RECIST。在一些實施方案中,受試者能夠表現出腫瘤標誌物,例如CA-125。在一個方面,與施用之前的CA-125水平相比,施用之後所述受試者中CA-125的表達水平被減少至少大約10%、至少大約20%、至少大約30%、至少大約40%、至少大約50%、至少大約55%、至少大約60%、至少大約65%、至少大約70%、至少大約75%、至少大約80%、至少大約85%、至少大約90%或至少大約100%。在一些實施方案中,受試者僅已經接受過一次針對復發性鉑敏感性疾病的基於鉑的治療,後續無鉑間隔不到六個月。在其它實施方案中,受試者具有美國東部腫瘤協作組(ECOG)體能狀況(performancestatus)0-1級。ECOG是用於評估患者的疾病進展、在患者的日常生活中疾病的作用和確定合適的治療和預後的量表或標準。表4顯示ECOG體能狀況:*如在Am.J.Clin.Oncol.:Oken,M.M.,Creech,R.H.,Tormey,D.C.,Horton,J.,Davis,T.E.,McFadden,E.T.,Carbone,P.P.:ToxicityAndResponseCriteriaOfTheEasternCooperativeOncologyGroup.AmJClinOncol5:649-655,1982中所出版的。在一些實施方案中,受試者具有與未患癌症的受試者可比的骨髓功能。與未患癌症的受試者可比的骨髓功能包括但不限於其作為主要造血器官和初級淋巴組織並且負責產生血細胞(例如紅細胞、粒細胞、單核細胞、淋巴細胞和血小板)的作用。骨髓結構和功能的詳細描述見於Jain,C.,1986b,Schalm’sVeterinaryHematology,Thehematopoieticsystem(LeaandFebiger,Philadelphia,PA),4,pp350–387;WeissandGeduldig,1991,Blood78:975–90;Wickramasinghe,1992,inHistologyforPathologists,Bonemarrow,edSternbergSS(RavenPress,NewYork),pp1–31;PickerandSiegelman,1999,inFundamentalImmunology,Lymphoidtissuesandorgans,edPaulWE(Lippincott-Raven,Philadelphia,PA),4,pp479–531;Hoffmanetal.,2000,HematologyBasicPrincipalsandPractice(ChurchillLivingstone,NewYork),3;AbboudandLichtman,2001,inWilliams』Hematology,Structureofthemarrowandthehematopoieticmicroenvironment,edsBeutlerE,LichtmanMA,CollerBS,KippsTJ,SeligsohnU(McGraw-Hill,NewYork),6,pp29–58,其每一篇通過引用整體併入本文。在進一步的實施方案中,受試者具有與未患癌症的受試者可比的血液學功能,其中血液學功能的指標選自由以下組成的組:a.中性粒細胞絕對計數(ANC)等於或高於1000/mm3;b.血小板(PLT)計數等於或高於100,000個/mm3;c.凝血酶原時間(PT)小於1.2x正常上限(ULN)秒;d.凝血活酶時間(PTT)小於1.2xULN秒,其中如果PTT高於ULN,則該患者狼瘡性抗凝集物質(LAC)為陰性;以及e.其任意組合。在其它實施方案中,受試者具有與未患癌症的受試者可比的器官功能,其中所述器官功能使用常見毒性標準進行分析,所述標準選自由以下組成的組:a.小於或等於1級常見毒性標準(CTC)的神經病;b.之前不超過30%的含有骨髓的主要區域已經接受過放射;c.小於2.5x正常上限(ULN)或小於5xULN的血清穀草轉氨酶(SGOT)、血清谷丙轉氨酶(SGPT)或鹼性磷酸酶;d.小於或等於1.5xULN水平的膽紅素;e.小於或等於1.5xULN水平的肌酐;f.在篩選時蛋白尿試紙小於2+或尿蛋白肌酐的比小於1.0;以及g.其任意組合。在又一些其它實施方案中,受試者已經從來自之前的治療(例如任何化療、放療或生物療法)的急性毒性中恢復。但是,所述受試者可能需要從1級神經病理、任意級的貧血或脫髮中恢復。在某些實施方案中,受試者之前已經接受過抗血管生成劑。在一些實施方案中,所述受試者之前沒有胃腸(GI)穿孔、GI梗阻或參與腸影像學研究。在又一些其它實施方案中,所述受試者沒有需要治療的活動的、未經治療的精神疾病或神經學症狀(允許有I級感覺性神經病理),不存在未經治療的中樞神經系統或腦轉移,沒有任何會阻止理解和/或給予知情同意的痴呆或顯著改變的精神狀況,沒有任何已知對聚氧乙烯蓖麻油(CremophorEL)的超敏反應,沒有不受控制的細菌、病毒或真菌感染的跡象,或其任意組合。在又一些其它實施方案中,如果所述受試者之前已經有紫杉醇反應,但隨後在重新攻擊時耐受該藥物,那麼所述受試者是適合本發明的。適合於本發明的受試者可以通過針對抗血管生成療法的血漿生物標誌物或細胞表面生物標誌物的測定進行鑑定。在一個實施方案中,適合於本發明的受試者表現出血漿生物標誌物,其包括但不限於血管內皮生長因子(VEGF)、磷脂醯肌醇聚糖生物合成F類蛋白(PIGF)、可溶性血管內皮生長因子受體-1(sVEGFR-1)、sVEGFR-2、sVEGFR-3、鹼性成纖維細胞生長因子(bFGF)、白細胞介素-1β(IL-1β)、白細胞介素-6(IL-6)、白細胞介素-8(IL-8)、腫瘤壞死因子-α(TNF-α)、vonWillerbrand因子(vWF)、可溶性c-kit(c-kit)、基質衍生因子1α(SDF1α)、甲胎蛋白(AFP)及其任意組合。在另一個實施方案中,適合於本發明的受試者表現出細胞表面生物標誌物,其包括但不限於CD31、CD34、CD45、CD133、血管內皮生長因子受體-1(VEGFR)、VEGFR-2或其任意組合。已知中和抗體的存在或形成在病毒的二次施用時可以阻礙有效基因轉移。在一個實施方案中,受試者沒有針對腺病毒的預先存在的抗體應答。在另一個實施方案中,在施用腺病毒時受試者沒有發展針對腺病毒的抗體應答。在本發明的一些方面,受試者沒有表現出狼瘡細胞抗凝集物質(LAC,也被稱為狼瘡細胞抗體、LA或狼瘡細胞抑制劑)。LAC的存在能夠通過任何已知的方法確定,例如通過由LAC測試或APLA測試測定所述LAC。LAC是結合與細胞膜有關的磷脂和蛋白質的免疫球蛋白。Joussen,J.,etal.,inDocumentaOphthalmologica,2008,Volume117,Number3,Pages263-265,RetinalVascularDisease,S.J.Ryan(eds),Springer,2007,780p,1040,ISBN:978-3-540-29541-9。LAC是促血栓形成劑;即,體內LAC抗體的存在沉澱血栓的形成。在實驗室試驗中這些抗體的存在引起aPTT的提高。據推測,所述抗體的存在幹擾了用來誘導體外凝聚的磷脂。據認為,體內與血小板膜磷脂發生相互作用,增加血小板的粘附和聚集;從而發揮其體內促血栓形成特徵。因此,LAC在體內充當凝血劑。四、表達FAS嵌合的腺病毒的均質群體本發明還提供包含SEQIDNO:19的腺病毒或具有ECACC保藏號13021201的腺病毒的均質群體。在本文中所使用的術語「均質」是指沒有汙染具有不同序列的異源腺病毒的腺病毒單個群體。異源腺病毒的實例包括但不限於包含含有SEQIDNO:20或SEQIDNO:21的核苷酸序列的腺病毒。包含含有SEQIDNO:20的核苷酸序列的腺病毒和包含含有SEQIDNO:21的核苷酸序列的腺病毒之前被公開於國際申請號PCT/IL2011/00007和PCT/IL2011/00009,其於2011年7月14日分別作為WO2011/083464和WO2011/083466出版,其通過引用整體併入本文。。本發明提供包含SEQIDNO:19(35207bp)的腺病毒,其包括不同於SEQIDNO:20(35203bp)和SEQIDNO:21的兩個核苷酸殘基。在SEQIDNO:20和SEQIDNO:21中的這兩個錯配(即Gly→Ala)位於核苷酸殘基第501位和第1255位。此外,SEQIDNO:19在3'端含有四個額外的鹼基對。另外,SEQIDNO:21含有編碼額外的E1區域的胺基酸。SEQIDNO:19包含在核苷酸殘基第458至1444位對應於SEQIDNO:18的內皮細胞特異性啟動子(即PPE-1-3x啟動子)的核苷酸序列,和在核苷酸殘基第1469至2569位對應於SEQIDNO:9的編碼FAS嵌合蛋白的核苷酸序列。在一個實施方案中,本發明是包含腺病毒的組合物,所述腺病毒包含被可操作地連接至內皮細胞特異性啟動子的FAS嵌合基因,其中所述腺病毒不含有SEQIDNO:20並且不含有SEQIDNO:21。在另一個實施方案中,本發明是包含含有SEQIDNO:19的腺病毒或具有ECACC保藏號13021201的腺病毒的組合物,其中所述組合物不含有包含SEQIDNO:20的腺病毒並且不含有包含SEQIDNO:21的腺病毒。在另一個實施方案中,組合物包含含有SEQIDNO:19的第458至2569位核苷酸殘基的腺病毒,其中所述組合物不包含含有SEQIDNO:20或SEQIDNO:21的第458至2569位核苷酸殘基的腺病毒。在其它實施方案中,本發明的組合物包含腺病毒,所述腺病毒包含與SEQIDNO:19至少80%、85%、90%、95%、96%、97%、98%、99%或100%相同的核酸序列,其中所述核酸序列不是SEQIDNO:20並且不是SEQIDNO:21,並且其中所述組合物不含有包含SEQIDNO:20的腺病毒並且不含有包含SEQIDNO:21的腺病毒。仍在一些其它實施方案中,本發明的組合物包含含有SEQIDNO:19的腺病毒或具有ECACC保藏號13021201的腺病毒,其中所述腺病毒是至少大約51%純的、至少55%純的、至少大約60%純的、至少大約70%純的、至少大約80%純的、至少大約90%純的、至少大約95%純的、至少大約96%純的、至少大約97%純的、至少大約98%純的、至少大約99%純的或大約100%純的。在本文中所使用的術語「純」是指均質性的程度,即沒有異源腺病毒。例如,包括包含SEQIDNO:19的腺病毒的大約51%純的組合物含有大約51%的包含SEQIDNO:19的腺病毒和大約49%的包含異源序列(例如SEQIDNO:20或SEQIDNO:21)的腺病毒。本發明的組合物可以含有其它成分,包括但不限於藥學可接受的載體、賦形劑、張力改性劑或用於製劑的任何必要成分。在一些實施方案中,本發明包括分離或純化所述核酸構建體的均質群體的方法。在其它實施方案中,本發明提供從包括包含SEQIDNO:19的腺病毒或具有ECACC保藏號13021201的組合物中去除腺病毒的異質群體(例如SEQIDNO:20、SEQIDNO:21)的方法。本發明還提供了在受試者中減少或降低腫瘤的尺寸或減緩腫瘤生長的速率、消除腫瘤的方法,該方法包括向所述受試者施用有效量的腺病毒的均質群體或包含腺病毒的均質群體的組合物。本發明還包括抑制、降低或減少腫瘤中新生血管形成或血管生成的方法,該方法包括向患有所述腫瘤的受試者施用有效量的腺病毒的均質群體或包含腺病毒的均質群體的組合物。另外,本發明包括在受試者中治療或預防與癌症有關或源自癌症的腫瘤的方法,該方法包括施用有效量的腺病毒的均質群體或包含腺病毒的均質群體的組合物。本發明還包括在受試者中減少或降低腫瘤的尺寸或減緩腫瘤生長的速率、消除腫瘤的方法,該方法包括向所述受試者重複施用有效量的腺病毒的均質群體或包含腺病毒的均質群體的組合物而不施用包含SEQIDNO:20或SEQIDNO:21的腺病毒的異質群體或包含腺病毒的異質群體的組合物。本發明還包括抑制、降低或減少腫瘤中新生血管形成或血管生成的方法,該方法包括向患有所述腫瘤的受試者重複施用有效量的腺病毒的均質群體或包含腺病毒的均質群體的組合物而不施用包含SEQIDNO:20或SEQIDNO:21的腺病毒的異質群體或包含腺病毒的異質群體的組合物。此外,本發明包括在受試者中治療或預防與癌症有關或源自癌症的腫瘤的方法,該方法包括重複施用有效量的腺病毒的均質群體或包含腺病毒的均質群體的組合物而不施用包含SEQIDNO:20或SEQIDNO:21的腺病毒的異質群體或包含腺病毒的異質群體的組合物。可以被用腺病毒的均質群體或組合物減少、抑制或治療的腫瘤可以是實體瘤、原發瘤或轉移。術語「轉移的」或「轉移」是指能夠在另一部位或器官建立繼發性腫瘤病灶的腫瘤細胞。在其它實施方案中,本發明的均質群體當被施用給需要其的受試者時延長該受試者的總體存活期。在進一步的實施方案中,本發明的均質群體當被施用給需要其的受試者時延長該受試者的無進展存活期。「實體瘤」包括但不限於肉瘤、黑色素瘤、癌或其它實體瘤癌症。「肉瘤」是指由像胚胎結締組織的物質組成並且通常由包埋於纖維狀或同源物質中的緊密堆積的細胞組成的腫瘤。肉瘤包括但不限於軟骨肉瘤、纖維肉瘤、淋巴肉瘤、黑色素肉瘤、黏液肉瘤、骨肉瘤、Abemethy's肉瘤、脂肉瘤、脂肪肉瘤、肺泡狀軟組織肉瘤、成釉細胞肉瘤、葡萄狀肉瘤、綠色瘤肉瘤、絨毛膜癌、胚胎性肉瘤、維爾姆斯瘤肉瘤、子宮內膜肉瘤、間質肉瘤、尤文肉瘤、筋膜肉瘤、成纖維細胞肉瘤、巨細胞肉瘤、粒細胞肉瘤、霍奇金肉瘤、特發性多發性色素沉著出血性肉瘤、B細胞的免疫母細胞肉瘤、淋巴瘤、T細胞的免疫母細胞肉瘤、Jensen's肉瘤、卡波西肉瘤、庫普弗細胞肉瘤、血管肉瘤、白色肉瘤、惡性間質瘤肉瘤、骨旁肉瘤、網狀細胞肉瘤、勞斯肉瘤、漿液囊性肉瘤、滑膜肉瘤和毛細管擴張性肉瘤。術語「黑色素瘤」是指由皮膚和其它器官的黑色素細胞系統產生的腫瘤。黑色素瘤包括例如肢端雀斑狀黑色素瘤、無黑色素性黑色素瘤、良性青少年性黑色素瘤、Cloudman's黑色素瘤、S91黑色素瘤、Harding-Passey黑色素瘤、青少年性黑色素瘤、惡性雀斑樣痣黑色素瘤、轉移性黑色素瘤、結節性黑色素瘤、甲下黑色素瘤(subungalmelanoma)和淺表擴散性黑色素瘤。術語「癌(carcinoma)」是指由傾向於滲透周圍組織且引起轉移的上皮細胞構成的惡性新生長。示例性的癌包括例如腺泡癌、腺泡狀癌、腺囊癌、腺樣囊性癌、癌性腺瘤(carcinomaadenomatosum)、腎上腺皮質癌、肺泡癌、肺泡細胞癌、基底細胞癌(basalcellcarcinoma)、基底細胞性癌(carcinomabasocellulare)、基底細胞樣癌、基底鱗狀細胞癌、細支氣管肺泡癌、細支氣管癌、支氣管癌、支氣管源性癌、腦形癌、膽管細胞癌、絨毛膜癌、膠樣癌、粉剌癌、子宮體癌、篩狀癌、encuirasse癌、皮膚癌、圓柱細胞癌(cylindricalcarcinoma)、柱狀細胞癌(cylindricalcellcarcinoma)、導管癌、硬癌、胚胎性癌、髓樣癌(encephaloidcarcinoma)、表皮樣癌(epiermoidcarcinoma)、腺樣上皮細胞癌、外植癌、exulcere癌、纖維癌、膠狀癌(gelatiniformcarcinoma)、膠質癌(gelatinouscarcinoma)、巨大細胞癌(giantcellcarcinoma)、巨細胞癌(carcinomagigantocellulare)、腺癌、粒層細胞癌、發基質癌(hair-matrixcarcinoma)、血樣癌、肝細胞癌、Hurthle細胞癌、膠樣癌(hyalinecarcinoma)、hypemephroid癌、幼稚型胚胎性癌、原位癌、表皮內癌、上皮內癌、Krompecher's癌、Kulchitzky細胞癌、大細胞癌、豆狀癌(lenticularcarcinoma)、豆狀癌(carcinomalenticulare)、脂肪瘤樣癌、淋巴上皮癌、髓樣癌(carcinomamedullare)、髓樣癌(medullarycarcinoma)、黑色素癌、軟癌(carcinomamolle)、粘液性癌(mucinouscarcinoma)、粘液癌(carcinomamuciparum)、粘液細胞癌(carcinomamucocellulare)、粘液表皮樣癌、粘液癌(carcinomamucosum)、粘液癌(mucouscarcinoma)、粘液瘤樣癌、鼻咽癌、燕麥細胞癌、骨化性癌(carcinomaossificans)、骨樣癌(osteoidcarcinoma)、乳頭狀癌、門脈周癌、浸潤前癌(preinvasivecarcinoma)、棘細胞癌、軟糊狀癌(pultaceouscarcinoma)、腎臟的腎細胞癌、備細胞癌(reservecellcarcinoma)、肉瘤樣癌、schneiderian癌、硬癌(scirrhouscarcinoma)、陰囊癌、印戒細胞癌、單純癌、小細胞癌、馬鈴薯狀癌、球狀細胞癌、梭形細胞癌、軟癌(carcinomaspongiosum)、鱗狀癌、鱗狀細胞癌、繩捆癌、血管擴張性癌(carcinomatelangiectaticum)、血管擴張性癌(carcinomatelangiectodes)、移行細胞癌、結節性皮癌(carcinomatuberosum)、結節性皮癌(tuberouscarcinoma)、疣狀癌和viflosum癌。可被抑制或治療的另外的癌(cancer)包括例如白血病、霍奇金病、非霍奇金淋巴瘤、多發性骨髓瘤、神經母細胞瘤、乳腺癌、卵巢癌、肺癌、橫紋肌肉瘤、原發性血小板增多症、原發性巨球蛋白血症、小細胞肺腫瘤、原發性腦瘤、胃癌、結腸癌、惡性胰腺胰島瘤、惡性類癌、膀胱癌、惡變前皮膚病變、睪丸癌、淋巴瘤、甲狀腺癌、乳頭狀甲狀腺癌、神經母細胞瘤、神經內分泌癌、食管癌、泌尿生殖道癌、惡性高鈣血症、宮頸癌、子宮內膜癌、腎上腺皮質癌、前列腺癌、米勒管癌、卵巢癌、腹膜癌、輸卵管癌或子宮乳頭狀漿液性癌。五、藥物組合物本發明中還提供了藥物組合物,其包含表達在本發明的方法中所使用的FAS嵌合體蛋白的核酸構建體,還提供了包含所述核酸構建體的腺病毒或所述腺病毒的均質群體。可以配製所述藥物組合物用於施用給哺乳動物,包括人類。在本發明的方法中所使用的藥物組合物包含藥學可接受的載體,包括例如離子交換劑、氧化鋁、硬脂酸鋁、卵磷脂、血清蛋白(如人血清白蛋白)、緩衝物質(如磷酸鹽)、甘氨酸、山梨酸、山梨酸鉀、飽和植物脂肪酸的偏甘油酯混合物、水、鹽或電解質、如硫酸魚精蛋白、磷酸氫二鈉、磷酸氫鉀、氯化鈉、鋅鹽、膠體二氧化矽、三矽酸鎂、聚乙烯吡咯烷酮、基於纖維素的物質、聚乙二醇、羧甲基纖維素鈉、聚丙烯酸酯、蠟、聚乙烯-聚氧丙烯-嵌段聚合物、聚乙二醇和羊毛脂。在一個實施方案中,所述組合物通過加入鹽水進行配製。本發明的組合物可以通過任何合適的方法進行施用,例如腸胃外(例如包括皮下、靜脈內、肌內、關節內、滑膜內、胸骨內、鞘內、肝內、病灶內和顱內注射或輸注技術)、心室內、口服、通過噴霧吸入、局部地、經直腸地、經鼻地、經頰地、經陰道地或經植入的貯庫。如之前所述,包括包含FAS嵌合體基因的核酸構建體或所述腺病毒的均質群體的組合物在內皮細胞中表達FAS嵌合體基因產物,並從而誘導該內皮細胞的細胞凋亡。因此,所述組合物能夠通過抑制腫瘤內皮細胞的新生血管形成和/或血管生成而抑制、減少或降低腫瘤或其轉移的尺寸。因此,在一個實施方案中,全身或局部遞送所述組合物。為了全身或局部遞送,含有所述核酸構建體、腺病毒或腺病毒的均質群體的藥物組合物可以利用機械裝置,如針、套管或手術器械。在本發明的方法中所使用的無菌可注射形式的組合物可以是水性或油性懸浮液。這些懸浮液可以根據本領域已知的技術使用合適的分散劑或溼潤劑和懸浮劑進行配製。該無菌可注射製劑還可以是在無毒腸胃外可接受的稀釋劑或溶劑中的無菌可注射溶液或懸浮液,例如在1,3-丁二醇中的懸浮液。可以使用的可接受的媒介物和溶劑包括水、林格氏溶液和等張氯化鈉溶液。此外,無菌的不揮發性油通常用作溶劑或懸浮介質。為了這個目的,任何溫和的不揮發性油都可以使用,包括合成的甘油單酸酯或甘油二酸酯。脂肪酸(如油酸及其甘油酯衍生物)可用於注射劑的製備,天然的藥學可接受的油如橄欖油或蓖麻油(尤其是它們的聚氧乙烯化的形式)也可以。這些油溶液或懸浮液也可以含有長鏈醇稀釋劑或分散劑,如羧甲基纖維素或在配製藥學可接受的劑型(包括乳劑和懸浮液)中常用的類似的分散劑。其它常用表面活性劑(如吐溫、司盤和在製備藥物可接受的固體、液體或其它劑型中常用的其它乳化劑或生物利用度增強劑)也可以用於配製的目的。腸胃外製劑可以是單次大丸藥劑量、輸注或負荷丸藥劑量接著維持劑量。這些組合物可以特定的固定或可變的時間間隔進行施用,例如每天一次或「按需」施用。在本發明的方法中所使用的某些藥物組合物可以可接受的劑型(例如膠囊、片劑、水性懸浮液或溶液)進行口服施用。某些藥物組合物還可通過鼻氣霧劑或吸入進行施用。這樣的組合物可以製備成在鹽水中的溶液,其中使用苄醇或其它合適的防腐劑、提高生物利用度的吸收促進劑和/或其它常規增溶劑或分散劑。可與載體材料組合以生產單個劑型的表達FAS嵌合體蛋白的核酸構建體、包含所述核酸構建體的腺病毒或所述腺病毒的均質群體的量將根據所治療的宿主、所使用的多肽類型和具體的施用模式而變化。組合物可以作為單個劑量、多個劑量或在所確立的時間段以輸注的形式進行施用。還可以調整劑量方案以提供所需最佳應答(例如治療性或預防性應答)。在一個實施方案中,包含表達FAS嵌合體蛋白的核酸構建體、腺病毒或所述腺病毒的均質群體的組合物在第1天進行輸注(即第一次劑量),並且接著一次或更多次後續劑量,例如,每一周的周期、每兩周的周期、每三周的周期、每四周的周期、每五周的周期、每六周的周期、每七周的周期、每八周的周期、每九周的周期、每十周的周期、每11周的周期或每12周的周期。在另一個實施方案中,包含表達FAS嵌合體蛋白的核酸構建體、腺病毒或所述腺病毒的均質群體的組合物在第1天進行輸注(即第一次劑量),並且接著一次或更多次後續劑量,例如,每兩周的周期、每個月的周期、每兩個月的周期、每三個月的周期、每四個月的周期、每五個月的周期或每六個月的周期。本發明的方法使用「有效量」或「治療有效量」的包含表達FAS嵌合體蛋白的核酸構建體、包含所述核酸構建體的腺病毒或所述腺病毒的均質群體的組合物。這樣的有效量或治療有效量可以根據如個體的疾病狀態、年齡、性別和體重的因素而變化。有效量或治療有效量可以是這樣的:其中治療有益效果勝過任何有毒或有害作用。針對任何具體患者的特定劑量和治療方案將取決於各種因素,包括所使用的具體組合物、所述患者的年齡、體重、一般健康狀況、性別和飲食、以及施用時間、排洩速率、藥物組合和要治療的具體疾病的嚴重程度。醫療護理人員對這樣的因素的判斷是在本領域普通技術範圍內的。量也將取決於要治療的個體、施用途徑、製劑類型、所使用的化合物的特徵、疾病的嚴重程度和所需效果。所用的量可通過本領域中公知的藥理學和藥代動力學原理來確定。包含編碼FAS嵌合體的核酸構建體的腺病毒或包含所述編碼FAS嵌合體的核酸構建體的腺病毒的均質群體的有效量可以是任何合適的劑量。在一個實施方案中,所述腺病毒或所述腺病毒均質群體的有效量是每個受試者至少大約109個VP、至少大約1010個VP、大約1011個VP、至少大約1012個VP或至少大約1013個VP。在另一個實施方案中,所述腺病毒或所述腺病毒均質群體的有效量是每個受試者至少大約1x1012個VP、至少大約2x1012個VP、大約3x1012個VP、至少大約4x1012個VP、至少大約5x1012個VP、至少大約6x1012個VP、大約7x1012個VP、至少大約8x1012個VP、至少大約9x1012個VP或至少大約1013個VP。在其它實施方案中,所述腺病毒或所述腺病毒均質群體的有效量是每個受試者大約109至大約1015個VP、大約1010至大約1014個VP、大約1011至大約1013個VP或大約1012至大約1013個VP或大約3x1012至大約1013個VP。在其它方面,如果第一次劑量沒有誘導任何毒性,則升高表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的有效量。例如,所述表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的第一次劑量可以是每個受試者至少大約1x109、大約1x1010個VP、至少大約1x1011個VP、至少大約1x1012個VP、至少大約2x1012個VP、至少大約3x1012個VP、至少大約4x1012個VP、至少大約5x1012個VP、至少大約6x1012個VP、至少大約7x1012個VP、至少大約8x1012個VP或至少大約9x1012個VP,並且所述表達FAS嵌合體蛋白的腺病毒的第二次劑量或後續劑量可以是每個受試者至少大約5x1012個VP、至少大約6x1012個VP、至少大約7x1012個VP、至少大約8x1012個VP或至少大約9x1012個VP、至少大約1x1013個VP、至少大約2x1013個VP、至少大約3x1013個VP、至少大約4x1013個VP、至少大約5x1013個VP、至少大約6x1013個VP、至少大約7x1013個VP、至少大約8x1013個VP、至少大約9x1013個VP、至少大約1x1014個VP。在一個具體的實例中,所述表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的第一次劑量是每個受試者大約3x1012個VP,並且所述表達FAS嵌合體蛋白的腺病毒的第二次劑量是每個受試者大約1x1013個VP。但是,如果一個特定的劑量誘導了劑量限制性毒性,則可以減少所述表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的有效量。包含所述腺病毒或所述腺病毒的均質群體的組合物可以向所述受試者輸注大約10分鐘、大約20分鐘、大約30分鐘、大約40分鐘、大約50分鐘、大約60分鐘、大約70分鐘、大約80分鐘、大約90分鐘、大約100分鐘、大約110分鐘或大約120分鐘。在一個實施方案中,本發明的所述核酸構建體被靜脈內輸注不超過60分鐘。在另一個實施方案中,包含所述腺病毒或所述腺病毒的均質群體的組合物以1mL/分鐘的速率、劑量為等於或小於3x1012個VP/受試者進行輸注。如果劑量大於3x1012個VP/受試者,則包含所述腺病毒或所述腺病毒的均質群體的組合物以1mL/分鐘的速率輸注第一個10mL,然後以3mL/分鐘的速率輸注剩餘的組合物。還可以將補充的活性化合物摻入本發明的方法中所使用的組合物。例如,編碼FAS嵌合體基因產物的核酸構建體或所述核酸構建體的均質群體可以與一種或更多種另外的治療劑共同配製和/或共同施用。本發明包括用於將編碼FAS嵌合體基因產物的核酸構建體或所述核酸構建體的均質群體遞送至選定靶組織的任何合適的遞送方法,包括推注水性溶液或植入控釋系統。控釋植入物的使用減少了重複注射的需要。編碼FAS嵌合體基因產物的核酸構建體、包含所述核酸構建體的腺病毒或所述腺病毒的均質群體可以被直接輸注進入腫瘤。用於化合物的直接腫瘤輸注的各種植入物是已知的,並且在治療化合物向患有女性婦科癌症的人類患者的遞送中是有效的。組合物還可以包含分散於作為化合物的合適遞送或支持系統起作用的生物相容性載體材料中的編碼FAS嵌合體基因產物的核酸構建體、包含所述核酸構建體的腺病毒或所述腺病毒的均質群體。緩釋載體的合適的實例包括成型顆粒形式(如栓劑或膠囊)的半透聚合物基質。可植入的或微膠囊的緩釋基質包括聚丙交酯(美國專利號3,773,319;EP58,481)、L-穀氨酸和γ-乙基-L-穀氨酸酯的共聚物(Sidmanetal.,Biopolymers22:547-56(1985));聚(2-羥乙基-甲基丙烯酸酯)、乙烯-乙酸乙烯酯(Langeretal.,J.Biomed.Mater.Res.15:167-277(1981);Langer,Chem.Tech.12:98-105(1982))或聚-D-(-)-3羥基丁酸(EP133,988)。根據本發明的方法,所述核酸構建體、所述腺病毒或所述腺病毒的均質群體的施用可以與一種或更多種化療劑的施用組合進行。該化療劑可以在表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的施用之前、同時或之後進行施用。可以與本發明的所述腺病毒共同施用的一種或更多種化療劑包括但不限於阿西維辛、阿柔比星、鹽酸阿考達唑、阿克羅寧、阿黴素、阿多來新、阿地白介素、愛寧達、六甲蜜胺、安波黴素、醋酸阿美蒽醌、氨魯米特、安吖啶、阿那曲唑、安特拉黴素、天冬醯胺酶、曲林菌素、阿扎胞苷、阿扎替派、阿佐黴素、巴馬司他、苯佐替派、比卡魯胺、鹽酸比生群、二甲磺酸雙奈法德、貝伐單抗、比折來新、硫酸博來黴素、布喹那鈉、溴匹立明、白消安、放線菌素C、卡魯睪酮、卡醋胺、卡貝替姆、卡鉑、卡莫司汀、鹽酸卡柔比星、卡折來新、西地芬戈、苯丁酸氮芥、西羅黴素、順鉑、克拉屈平、甲磺酸克立那託、環磷醯胺、阿糖胞苷、達卡巴嗪、放線菌素D、鹽酸柔紅黴素、地西他濱、右奧馬鉑、地扎呱寧、甲磺酸地扎呱寧、地吖醌、多西他賽、多柔比星、鹽酸多柔比星、屈洛昔芬、檸檬酸屈洛昔芬、丙酸甲雄烷酮、達佐黴素、依達曲沙、鹽酸依氟鳥氨酸、依沙蘆星、恩洛鉑、恩普氨酯、依匹哌啶、鹽酸表柔比星、厄布洛唑、鹽酸依索比星、雌莫司汀、雌莫司汀磷酸鈉、依他硝唑、依託泊苷、磷酸依託泊苷、氯苯乙嘧胺、鹽酸法倔唑、法扎拉濱、芬維A胺、氟尿苷、磷酸氟達拉濱、氟尿嘧啶、氟西他濱、磷喹酮、磷曲星鈉、吉西他濱、鹽酸吉西他濱、羥基脲、鹽酸伊達比星、異環磷醯胺、伊莫福新、幹擾素α-2a、幹擾素α-2b、幹擾素α-nl、幹擾素α-n3、幹擾素β-Ia、幹擾素γ-Ib、羥丙氨鉑、鹽酸伊立替康、醋酸蘭瑞肽、來曲唑、醋酸亮丙瑞林、鹽酸利阿唑、洛美曲索鈉、洛莫司汀、鹽酸洛索蒽醌、馬索丙考、美登素、鹽酸氮芥、醋酸甲地孕酮、醋酸美侖孕酮、美法侖、美諾立爾、巰嘌呤、甲氨喋呤、甲氨喋呤鈉、氯苯氨啶、美妥替哌、米丁度胺、米託凱星、絲裂紅素、米託菌素、米託馬星、絲裂黴素、米託司培、米託坦、鹽酸米託蒽醌、麥考酚酸、諾考達唑、諾拉黴素、奧馬鉑、奧昔舒侖、帕唑帕尼(pazotinib)、紫杉醇、培門冬酶、培利黴素、溴新斯的明、硫酸培洛黴素、過磷醯胺、哌泊溴烷、哌泊舒凡、鹽酸吡羅蒽醌、普卡黴素、普洛美坦、卟吩姆鈉、泊非黴素、潑尼莫司汀、鹽酸丙卡巴肼、嘌羅黴素、鹽酸嘌羅黴素、吡唑呋林、利波腺苷、羅谷亞胺、沙芬戈、鹽酸沙芬戈、司莫司汀、辛曲秦、索拉非尼(Sorafinib)、司巴膦酸鈉、司帕黴素、鹽酸鍺螺胺、螺莫斯汀、螺鉑、絳色黴素、鏈佐星、磺氯苯脲、舒尼替尼(Sunitinib)、他利黴索、紫杉酚、替可加蘭鈉(tecogalansodium)、替加氟、鹽酸替洛蒽醌、替莫泊芬、替尼泊苷、替羅昔隆、睪內酯、硫咪嘌呤、硫鳥嘌呤、噻替派、噻唑呋林(Tiazofuirin)、替拉扎明、鹽酸拓撲替康、檸檬酸託瑞米芬、醋酸曲託龍、磷酸曲西立濱、三甲曲沙、三甲曲沙葡萄糖醛酯、曲普瑞林、鹽酸妥布氯唑、尿嘧啶氮芥、烏瑞替派、伐普肽、維替泊芬、硫酸長春鹼、硫酸長春新鹼、長春地辛、硫酸長春地辛、硫酸長春匹定、硫酸長春甘酯、硫酸環氧長春鹼、長春瑞濱、硫酸異長春鹼、硫酸長春利定、伏氯唑、折尼鉑(Zeniplatin)、淨司他丁或鹽酸佐柔比星。另外的抗腫瘤劑包括在抗腫瘤劑(PaulCalabresi和BruceA.Chabner)第52章及其引言、Goodman和Gilman的"ThePharmacologicalBasisofTherapeutics",EighthEdition,1990,McGraw-Hill,Inc.的第1202-1263頁中所公開的那些。在一些實施方案中,施用表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的受試者同時進行放療治療。在其它實施方案中,施用表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的受試者用兩種或更多種化療劑同時進行治療。在某些實施方案中,施用表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的受試者同時用化療劑和放療進行治療。在本發明的組合療法方面,化療劑可以是紫杉醇。在一個方面,表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體可以與紫杉醇同時施用。在另一個方面,表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體可以在紫杉醇的施用之前或之後施用。在其它實施方案中,紫杉醇在施用表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體之前至少30分鐘、至少大約一小時、至少大約兩小時、至少大約三小時、至少大約四小時、至少大約五小時、至少大約六小時、至少大約七小時、至少大約八小時、至少大約九小時、至少大約十小時、至少大約11小時、至少大約12小時、至少大約13小時、至少大約14小時、至少大約19小時、至少大約20小時、至少大約21小時、至少大約22小時、至少大約23小時、至少大約24小時、至少大約一天、至少大約36小時、至少大約2天、至少大約60小時或至少大約3天進行施用。化療劑還可以任何合適的方法進行施用,例如胃腸外、心室內、口服、通過吸入噴霧、局部地、經直腸地、經鼻地、經頰地、經陰道地或經植入的貯庫。在本文中所使用的術語「腸胃外」包括皮下、靜脈內、肌內、關節內、滑膜內、胸骨內、鞘內、肝內、病灶內、腹膜內、顱內注射或輸注技術。化療劑的有效量可由本領域獲得。在一個方面,例如,紫杉醇的有效量可以是至少大約10mg/m2、至少大約20mg/m2、至少大約30mg/m2、至少大約40mg/m2、大約50mg/m2、至少大約60mg/m2、至少大約70mg/m2、至少大約80mg/m2、大約90mg/m2、至少大約100mg/m2或至少大約110mg/m2。在另一個方面,紫杉醇的有效量是從大約10mg/m2至大約200mg/m2、從大約20mg/m2至大約150mg/m2、從大約30mg/m2至大約100mg/m2或從大約40mg/m2至大約80mg/m2。在其它方面,紫杉醇的有效量是大約10mg/m2、大約20mg/m2、大約30mg/m2、大約40mg/m2、大約50mg/m2、大約60mg/m2、大約70mg/m2、大約80mg/m2、大約90mg/m2或大約100mg/m2。在紫杉醇施用的某些方面,紫杉醇被輸注至少10分鐘、至少20分鐘、至少30分鐘、至少40分鐘、至少50分鐘、至少60分鐘、至少70分鐘、至少80分鐘、至少90分鐘、至少100分鐘、至少110分鐘、至少120分鐘、至少150分鐘、至少180分鐘、至少210分鐘、至少240分鐘、至少270分鐘或至少300分鐘。在特定的實例中,紫杉醇至少進行一小時輸注。可以使用本領域已知的任何方法用於紫杉醇的輸注方法。例如,可以通過具有不大於0.22微米的微孔膜的管線濾器(inlinefilter)施用紫杉醇超過三小時。在一些方面,紫杉醇在第1天(施用表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體的同一天)進行輸注(即第一次劑量),並且接著一次或更多次後續劑量,例如,每三天、每四天、每五天、每六天、每七天、每八天、每九天、每十天、每11天、每12天、每13天或每14天輸注。在一個特定的實例中,在每一個28天中,紫杉醇在第1天施用(即第一次劑量)並且接著在第8天(即第二次劑量)、第15天(即第三次劑量)和第22天(即第四次劑量)施用。在又一些其它方面,如果第一次劑量沒有誘導任何毒性,則升高要與表達FAS嵌合體蛋白的腺病毒或所述腺病毒的均質群體共同施用的化療劑的有效量。例如,紫杉醇的第一次劑量可以是大約10至大約70mg/m2、大約20至大約60mg/m2、大約30至大約50mg/m2或大約40mg/m2,並且第二次劑量或任何後續劑量可以提高大約10mg/m2、大約20mg/m2、大約30mg/m2、大約40mg/m2、大約50mg/m2、大約60mg/m2、大約70mg/m2或大約80mg/m2。在一個實施方案中,紫杉醇的第一次劑量是大約40mg/m2,紫杉醇的第二次劑量是大約80mg/m2。在另一個實施方案中,紫杉醇的第一次劑量是大約40mg/m2,紫杉醇的第二次劑量是大約80mg/m2,紫杉醇的第三次劑量是大約80mg/m2。在其它實施方案中,紫杉醇的第一次劑量是大約40mg/m2,紫杉醇的第二次劑量是大約80mg/m2,紫杉醇的第三次劑量是大約80mg/m2,紫杉醇的第四次劑量是大約80mg/m2。但是,如果第一次劑量對受試者誘導劑量限制性毒性,則可以減少紫杉醇的有效量。可以通過任何已知方法確定該劑量限制性毒性:例如,(1)中性粒細胞絕對計數(ANC)<0.5x109/L持續≥4天或者中性粒細胞絕對計數100.2°F);(2)持續任意時間的血小板計數<10x109/L;(3)任何其它藥物有關的非肝和非血液學≥3級的毒性,或其任意組合。這不包括可被醫學控制的≥3級的噁心或嘔吐(如果噁心和/或嘔吐不能被醫學控制並且在第一周期期間發生,這將被認為是DLT)或≥3級的低鉀血症、低鈉血症、低磷血症、低鎂血症、低鈣血症,如果能被很容易地校正,在臨床上是無症狀的,並且沒有伴隨醫學顯著併發症(例如EGC變化)。在一個實施方案中,在第1天的紫杉醇的第一次劑量是40mg/m2,且如果受試者沒有表現出劑量限制性毒性,則第8天的第二次劑量和後續第15天、第22天和第28天的劑量提升至80mg/m2。在另一個實施方案中,如果受試者中ANC/藥物≥1,000/mm3,則對所述受試者施用完全劑量的紫杉醇,例如80mg/m2。如果受試者中ANC/藥物<1,000/mm3,則停止紫杉醇的施用。如果進一步發生ANC<1,000/mm3,則紫杉醇的劑量可以減少至60mg/m2。在其它實施方案中,如果受試者具有血小板/藥物≥100,000/mm3或第一次發生75,000/mm3,則所述受試者施用完全劑量的紫杉醇,例如80mg/m2。如果受試者顯示<75,000/mm3的血小板/藥物,則停止紫杉醇的施用。如果受試者顯示重複發生血小板/藥物75,000/mm3,則紫杉醇的劑量降至69mg/m2。在一些實施方式中,如果受試者表現出第一次發生神經病等級>2,則紫杉醇的劑量從全劑量(例如大約80mg/m2)減少至大約60mg/m2。如果受試者表現出第二次發生神經病等級>2(或儘管劑量減少仍顯示該病頑固存在),則紫杉醇劑量減少至40mg/m2。如果受試者表現出第三次發生(或儘管劑量減少但該病仍保持),則中止施用紫杉醇。尚不清楚紫杉醇會引起肝毒性;但是,它在具有嚴重肝功能障礙的患者中的清除被延遲。因此,具有肝功能障礙的受試者中的紫杉醇劑量可以根據下表進行修改。表5.針對具有肝功能障礙的受試者的紫杉醇劑量修改血清AST或ALT紫杉醇1.5mg/dl的受試者直到該異常實驗室值改善至≤1級才可以接受紫杉醇。按照以上表格,在恢復後治療可以重新開始,紫杉醇為低於一個劑量水平。表現出紫杉醇毒性持續超過2周的受試者可以中止該藥物。在某些實施方案中,受試者在施用一種或更多種化療劑(例如紫杉醇)之前、同時或之後施用免疫抑制劑。在一個實施方案中,可用於施用給受試者的免疫抑制劑選自由以下組成的組:H2-受體拮抗劑、西咪替丁、雷尼替丁、法莫替丁、皮質類固醇、地塞米松、環孢黴素、苯海拉明及其任意組合。在進一步的實施方案中,本發明的方法還包括在施用表達FAS嵌合體蛋白的核酸構建體之前、同時或之後施用一種或更多種抗血管生成劑。在一些實施方案中,本發明的受試者被施用止吐劑。合適的止吐劑的實例包括但不限於5-HT3受體拮抗劑(例如多拉司瓊、格拉司瓊、昂丹司瓊、託烷司瓊、帕洛諾司瓊或米塔扎平)、多巴胺拮抗劑(例如多潘立酮、奧氮平、氟哌啶、氟哌啶醇、氯丙嗪、異丙嗪、丙氯拉嗪、甲氧氯普胺、阿立必利或丙氯拉嗪(compazine、stemzine、buccastem、stemetil或phenotil)、NK1受體拮抗劑(例如阿瑞吡坦(aprepitant)或卡索吡坦(casopitant))、抗組胺劑(H1組胺受體拮抗劑)(例如賽克利嗪、苯海拉明、茶苯海明、多西拉敏、美克洛嗪、異丙嗪或羥嗪)、大麻類物質(例如大麻、屈大麻酚、大麻隆、JWH系列或四氫大麻酚(Sativex))、苯二氮類(例如咪達唑侖或蘿拉西泮)、抗膽鹼藥(例如東莨菪鹼)、類固醇(例如地塞米松)、曲美苄胺、姜、愈吐寧錠(emetrol)、異丙酚、薄荷、蠅蕈醇或ajwain、或其任意組合。在其它實施方案中,合適的止吐劑是地塞米松、PRN蘿拉西泮(Ativan)、格拉司瓊(kytril)、丙氯拉嗪(compazine)、奮乃靜、昂丹司瓊(zofran)、奮乃靜或其任意組合。在其它實施方案中,在施用編碼FAS嵌合體蛋白的核酸構建體之前、同時或之後施用解熱藥。解熱藥的實例包括但不限於NSAIDs(例如布洛芬、萘普生、酮洛芬和尼美舒利)、阿司匹林、對乙醯氨基酚、安乃近、萘丁美酮、安替比林、奎寧或其任意組合。本發明的方法還包括在接受表達FAS嵌合體蛋白的核酸構建體或所述化療劑之前或之後確定受試者中的疾病進展。在一個實施方案中,通過測定腫瘤的尺寸確定疾病進展。在另一個實施方案中,通過測定腫瘤抗原(例如CA-125)的表達確定疾病進展。為了確定所述疾病進展,在表達FAS嵌合體基因產物的核酸構建體的輸注之前、在所述輸注結束時、所述輸注之後大約三小時和/或所述輸注之後大約六小時收集受試者的血液和尿液。在其它實施方案中,在0分鐘-15分鐘(就在給藥之前)、給藥開始之後30分鐘±15分鐘、給藥開始之後60分鐘±15分鐘、給藥開始之後四小時±15分鐘、給藥開始之後六小時±15分鐘和/或在任何不良事件發生時記錄受試者的生命體徵。要測定的生命體徵包括但不限於收縮和舒張壓、外周心率、體溫、呼吸率或其任意組合。在又一些其它實施方案中,測定受試者的血液學(例如全血細胞分類計數、INR和活化的PTT)、凝固(例如PTT水平)、表達FAS嵌合體的核酸構建體的生物分布、血管生成性和炎症性生物標誌物的表達(例如VEGF、PlGF、sVEGFR1、bFGF、IL-1β、IL-6、IL-8、TNF-α、sVEGFR2、SDF1α、CD31、CD34、VEGFR2、vWF、其任意組合)、腫瘤標誌物(例如CA-125)的表達或其任意組合。對於發熱和嗜中性粒細胞減少症,以常規方式管理適合於本發明方法的受試者。在重新開始療法之前,受試者需要從發熱和活躍性感染問題中恢復。在一些實施方式中,不使用生長因子的受試者在其下一個周期中將添加Neupogen或Neulasta。接受Neupogen5μg/kg/天的受試者可以在同一方案內將他們的劑量升高至10μg/kg。實施例實施例1病毒載體的構建和克隆所述載體使用含有大部分5型腺病毒基因組的骨架構建,並與銜接質粒部分同源,從而能夠進行重組。從骨架質粒上去除E1早期轉錄單位,並且缺失pWE25和Amp抗性選擇標記位點而對其進一步修飾。銜接質粒(含有Ad5、CMV啟動子、MCS和SV40polyA序列)通過限制性消化去除其中的CMV啟動子,並插入PPE-1啟動子和Fas-c片段對其進行修飾。修飾的PPE-1啟動子(PPE-1-3X,SEQIDNO:18)和Fas嵌合轉基因(Fas-c,SEQIDNO:9)被用來構建腺病毒載體。PPE-1-(3X)-Fas-c元件(2115bp)由PPE-1-(3X)-luc元件構建而來。該元件含有1.4kb的小鼠前內皮素原PPE-1-(3X)啟動子、螢光素酶基因、SV40polyA位點和小鼠ET-1基因的第一個內含子,其來自Harats等人使用的pEL8質粒(8848bp)(HaratsD.等,JCI,1995)(HaratsD.etal.,JCI,1995)。使用BamHI限制性酶從pEL8質粒中獲取PPE-3-Luc盒。螢光素酶基因被Fas-c基因[由人TNF-R1(腫瘤壞死因子受體1,SEQIDNO:4)的胞外和膜內結構域以及Fas(p55)胞內結構域(SEQIDNO:8)組成(Boldin等,JBC,1995)]取代而獲得PPE-l-3x-Fas-c盒。PPE-1(3x)-Fas-c質粒–所述盒通過限制性消化被進一步引入到骨架質粒中,得到PPE-1(3x)-Fas-c質粒。銜接子-PPE-l(3x)-Fas-c質粒–從第一代構建體PPE-1-3x-Fas-c質粒中提取PPE-l-3x-Fas-c元件,並使用選定的PCR引物進行擴增,分別在5』-和3』-末端引入SnaB1和EcoR1限制性位點。利用這些位點將PPE-Fas-c片段克隆進經SnaB1和EcoR1消化的銜接質粒,獲得銜接子-PPE-l-3x-Fas-c質粒,用於宿主細胞的轉染(例如,PER.C6細胞)。實施例2VB-111在具有轉移性Lewis肺癌(LLC)的小鼠中的效力和安全性總結在本研究中,使用VB-111(109或1011個病毒顆粒(VPs))、卡鉑(Carboplatin)(20或50mg/kg)和愛寧達(Alimta)(10或30mg/kg)或這些小分子和腺病毒載體的組合處理LLC模型小鼠。使用兩個劑量的VB-111的處理都獲得了100%存活。施用低劑量的化療劑獲得輕微降低的存活率(94.1%),其與施用媒介物看到的相似(93.8%)。但是,施用高劑量的化療劑導致最低的存活百分比(50%)。與單獨施用VB-111相比,組合治療降低了存活率(64%-93%)。在不同組中器官重量(心臟、腎臟和腦)基本相同。不同組之間在肝、脾和睪丸重量上發現一些不同,主要是單獨高劑量VB-111或與該劑量的任何組合的重量較高。與媒介物處理相比,在肝功能上沒有發現顯著差異。通常,組合療法沒有比組成其的單一療法引起更高的毒性。VB-111109+高化療劑的組合比單獨的高化療劑處理明顯更有效。另外,組合處理提高了VB-111109單獨處理的平均和中值腫瘤負荷(分別是1.5倍平均值和5倍的中值),雖然統計學上不顯著。如果在更大組上應用該處理,這種效果可能會呈現統計學上的顯著性。VB-111109+高化療劑的組合獲得了與VB-1111011類似的高腫瘤負荷減輕,因為在這兩組之間沒有明顯的差異。單獨VB-111109的處理沒有獲得這種與VB-1111011的明顯相似性。VB111109+高化療劑組合療法可能會降低VB-111的劑量,卻保持其高效力,並改善卡鉑和愛寧達單獨化療的低效力。介紹Lewis肺癌(LLC)是一種廣泛應用於轉移的小鼠模型(Varda-Bloom等,2001)。本研究將要確定VB-111與已經明確的化療法(例如卡鉑和愛寧達)組合治療時的可能的協同性:評價單一劑量VB-111(109或1011VPs/小鼠)在單獨處理或與卡鉑(20或50mg/kg)和愛寧達(10或30mg/kg)重複腹膜內處理組合時的效力,並評價和鑑定VB-111單獨處理和與上述的化療劑組合時的安全性和耐受性。表6顯示了本研究設計。表6.研究設計材料和方法測試和參考材料名稱:VB-111(PPE-fas).化學名稱:未說明活性組分:5型腺病毒,E1缺失,E3部分缺失,具有PPE-1(3x)啟動子,含有fas嵌合轉基因媒介物:PBS和甘油提供:VBL物理狀態:液體保存條件:≤-65℃,低溫瓶中物品準備:處理當天融化藥瓶並顛倒混合名稱:卡鉑化學名稱:Ebewe媒介物:注射用水提供:Ebewe物理狀態:液體(450mg/45ml)保存條件:RT名稱:愛寧達化學名稱:Pemetrexed媒介物:注射用水提供:Lilly物理狀態:粉末保存條件:RT對照品:名稱:PBS10%甘油提供:VBL物理狀態:液體保存條件:≤-65℃,低溫瓶中.物品準備:處理當天融化藥瓶並置於冰上不超過30分鐘。融化D122Lewis肺癌細胞。在細胞注射的第一天,收集細胞並懸浮至終濃度5x105/50μl,注射至左足墊。在注射後5天用卡尺測量腫瘤的直徑。接下來每周測量直至其達到5mm,然後每天測量直至7mm時切除(定義為第-5天)。處死後,收集腦、心臟、肝臟、脾臟、腎臟和睪丸,稱重並根據下列參數評估:整個器官的顏色和質地結構、病灶的存在或器官內轉移的任何證據(切片後)。肝功能實驗室分析(血液中GOT和GPT水平)包括在第5±1天和研究結束時(第16天)取自每組5隻小鼠的樣品。所有的動物程序都經過Tel-Hashomer的Sheba醫學中心的「動物護理與使用委員會」(「AnimalCareandUseCommittee」ofShebaMedicalCenter,Tel-Hashomer)的批准。本研究為有安慰劑對照的盲研究。C57Bl6小鼠通過左足墊皮下注射接受Lewis肺癌細胞(D122)。當腫瘤組織達到直徑7mm時(大約細胞注射後3周),在麻醉下將具有原發腫瘤的足墊切除。將這一天定義為第-5天。原發腫瘤切除後5天、第一次施用日為第0天。在第0天將小鼠隨機分為如下表所述的不同處理組。將測試的腺病毒載體或對照物質進行尾靜脈注射,每隻小鼠的總體積為100μl。IP施用卡鉑和愛寧達,每隻小鼠每天劑量為總體積100μl。在整個研究過程中直至處死時,針對安全性和效力對動物進行跟蹤(如下面評估規劃表所列)。記錄研究中每隻動物的死亡,並試圖確定死亡原因。對每隻動物的處死日設定如下:當第5隻對照小鼠(PBS10%甘油–組9)死於轉移時,確定從第0天(對該小鼠第一次IV施用媒介物)開始經過的天數。該天數設定為所有仍存活小鼠的處死日(即如果第5隻對照小鼠在其第16天死亡,那麼每隻小鼠都在到達其自己的第16天時被處死)。效力評估規劃示於表7中。表7.評估規劃*皮膚、皮毛、眼睛、黏膜、呼吸、神經控制、震顫、流涎、腹瀉、活動、昏睡、抽搐、異常行為模式、睡眠、昏迷。**來自每組5隻小鼠結果在單獨用109和1011VPsVB-111處理的小鼠組中,至第16天時所有動物都存活。組合治療的存活率降低,而最低的存活率見於用高劑量的化療劑處理的小鼠中,如表8所示。表8.第16天的死亡率。為了評估效力,在處死日稱重肺。正常肺質量為~0.2g。腫瘤負荷定義為肺質量減去0.2g。平均和中值腫瘤負荷示於表9中。表9.平均和中值腫瘤負荷與媒介物處理相比導致降低的腫瘤負荷的所有處理示於圖1中。圖1顯示,使用E11、E9+高化療劑和E11+高化療劑的處理比其它處理組顯示更低的平均腫瘤負荷。對效力進行了幾種統計學比較。當使用單因素方差分析時,媒介物處理比除了VBE9+低化療劑組合外的所有其它處理具有更高的腫瘤負荷,這具有統計學上的顯著性(p=18.2.組織學上確定的卵巢上皮、腹膜或輸卵管癌症,以及子宮乳頭狀漿液性癌(UPSC)和婦科MMMT。3.允許多達3條以前的轉移性疾病化療線。4.患者必須之前進行過鉑或基於鉑的化療。5.EasternCooperativeOncologyGroup(ECOG)狀態0-1.6.在完成或正在接受含有鉑和紫杉烷的療法的6個內,根據成像或CA-125定義鉑抗性或難治療性疾病為進展性疾病(原發或繼發-即對於復發性鉑敏感性疾病,患者可能只接受一次基於鉑的治療,接下來無鉑間隔<6個月)。7.需要使用RECIST或CA-125可測量的或可評估的疾病(對於標準化資格,如果一些患者根據RECIST和CA-125都符合,則RECIST優先)。8.適當的骨髓功能。9.適當的血液學功能:i.ANC≥1000/mm3ii.PLT≥100,000/mm3iii.PT和PTT(秒數)ULN的受試者必須具有陰性的LAC)10.適當的器官功能:a.CTC神經病學低於或等於1級。b.之前的放射必須沒有包括≥30%的含有主要骨髓的區域(骨盆、腰椎)c.SGOT/SGPT/鹼性磷酸酶≤2.5xULN或<5xULN(對於記載的肝轉移)d.膽紅素≤1.5xULNe.肌酸酐≤1.5xULNf.篩選時浸棒的蛋白尿<2+或UPC(尿蛋白肌酸酐比1/2茶勺的鮮紅的血)。15.出血素質或凝血功能明顯異常的跡象(沒有抗凝治療)。16.在第一天前的28天內有大的手術操作、開放性活檢或重大創傷性病變,或者預計在研究過程中需要大的手術操作。17.在第一天之前的7天內進行了中心活檢或其它小的外科手術操作,不包括放置血管通路裝置。18.在第一天前的6個月內腹壁瘻或胃腸穿孔病史。19.當前的腸梗阻指徵和症狀。20.當前的對IV水合或腸道外全營養(TPN)的依賴性。21.嚴重的不癒合的傷口、活躍的潰瘍或未治療的骨折。22.對於TKI在前面4周內或對於抗體或基於肽抗體(peptibody)的治療6周內接受抗血管生成治療的患者。23.持續需要免疫抑制治療的患者,包括使用環孢菌素,或具有在列入前最後4周內長期使用任何此類藥物的歷史的患者。允許穩定劑量的皮質類固醇(例如在施用前至少2周開始的)。組成:AD5-PPE-L-3X-FAS-嵌合體Ad5-PPE-l-3X-fas-嵌合體(SEQIDNO:19)是一種血管破壞性基因治療劑,由含有修飾的小鼠前內皮素原啟動子(PPE-l-3x,SEQIDNO:18)和fas-嵌合轉基因[Fas和人腫瘤壞死因子(TNF)受體](SEQIDNO:9)的非複製性腺病毒載體(Ad5,E1缺失的)組成。其配製成無菌載體溶液,並在一次性使用的小瓶中以冷凍形式提供(低於-65℃)。每個小瓶含有1.1mL的具有特定病毒滴度的載體溶液。效力和藥效學研究目的:治療計劃用Ad5-PPE-l-3X-fas-嵌合體與每周一次的紫杉醇組合處理患者。周期長度為28天。患者將每周(第1、8、15和22天)接受60分鐘IV輸注的紫杉醇。在周期1開始的奇數周期的第一天經IV輸注施用Ad5-PPE-l-3X-fas-嵌合體,之後每兩個周期進行一次。處理計劃示於圖3。階段I的劑量水平示於下列表10。表10:階段I1部分的劑量水平:腳註:1.在確定最大耐受劑量(MTD)之後或劑量水平3已經完成後轉換到階段II。2.請注意,對於<50kg的受試者,在各個劑量水平時總VB-111病毒顆粒劑量與所示的不同。3.對於劑量水平1計劃在受試者內進行劑量逐漸升高。在周期2第一天,對列入到劑量水平1的參與者,紫杉醇的劑量可能被升高至80mg/m2,考慮到這是臨床使用的標準劑量。如果兩名參與者在劑量水平1經歷劑量限制性毒性(DLT),則終止該研究,不確定MTD。4.這些數字可反應包括有:(a)直接列入到劑量水平2的參與者,以及(b)劑量水平1參與者,其已在周期2的第一天升高劑量至劑量水平2,用於評價周期3過程中的DLT。階段I組群的參與者將因此通過修飾的3+3設計被列入,如表11所示和如下所述。從劑量水平1升高劑量至劑量水平2:如果2名參與者在劑量水平1經歷DLT,研究將終止,不確定MTD。在劑量水平1中,計劃在周期2的第一天,在受試者內將參與者劑量升高至80mg/m2的紫杉醇,然後這些參與者可以被算作評價列入劑量水平2的參與者,儘管VB-111和紫杉醇同時施用發生在劑量水平1的周期3(即列入劑量水平2的參與者和具有紫杉醇的劑量升高至80mg/m2的周期3劑量水平1的參與者都將被評價,所以在劑量水平2可有2-12名參與者,其中多至6名起始列入劑量水平1,然後劑量被升高)。如果在評價了3名受試者後在劑量水平2沒有DLT(或者作為劑量水平2的新參與者或者由於劑量水平1中的紫杉醇劑量升高),該研究將列入劑量水平3。該計劃只適用於在受試者內紫杉醇的劑量升高(以使進行40mg/m2劑量的參與者數最小化),沒有VB-111的受試者內劑量升高。從劑量水平2到劑量水平3的劑量升高:一旦3名受試者列入劑量水平2,在起始施用VB-111後觀察14天,沒有DLT,則受試者可列入劑量水平3(1x1013VPs),因為至少6名參與者將已經在劑量水平1和2被施用3x1012VPs。只有當至少2名患者接受一劑施用3x1012VPs而沒有DLT,才可在劑量水平3第二次施用VB-111。另外,將在平行的VB-111I/II期研究的同時進行DLT監測。從這些研究得到的安全性發現將在該團隊中分享。如果在劑量水平3發生任何DLT,參與者可繼續研究,並將接受進一步施用劑量水平2的VB-111(3x1012VPs)。表11:劑量升高決定規則鑑於藥物的新穎性,將在開始研究的每名新人之間至少間隔2天按順序列入新參與者。多個新研究的參與者不會在同一天施用。一個28天的處理周期的完成將作為確定每一施用組群的MTD和DLT的基礎。將根據CommonTerminologyCriteriaforAdverseEvents(CTCAE)第4版(在國際網際網路:http://ctep.info.nih.gov有可下載文件)來評價毒性等級。定義劑量限制性毒性(DLT)對於DL1參與者,將在第一周期(周期1)和第三周期評估潛在的DLT。將通過CTCAE4.0版來評估毒性。劑量限制性毒性定義如下:1.中性粒細胞絕對計數<0.5x109/L持續≥4天,或者中性粒細胞絕對計數100.2°F),其不容易用解熱藥控制。2.血小板計數<10x109/L持續任何時間。3.任何其它藥物有關的非肝和非血液系統等級≥3的毒性。這不包括等級≥3的可以被藥物控制的噁心或嘔吐(如果噁心和/或嘔吐不能被藥物控制並發生在第一周期,它將被認為DLT)或等級≥3的可以被容易矯正的、臨床無症狀的、並不伴有醫學上嚴重併發症(例如ECG改變)的低鉀血症、低鈉血症、低磷血症、低鎂血症和低鈣血症。發生在施用VB-111後24小時內的等級3-4的發熱事件,如果他們對有症狀的治療有反應,則不被認為是DLT。在任何時間點,如果患者有不可接受的毒性和/或進展,其將被移出研究。研究過程預處理評估:臨床醫師(MD或NP)將評估患者是否滿足合格標準。在開始第一個處理周期處理之前的14天內將進行病史、身體檢查,並記錄重量和生命指徵以及記錄ECG。建立基線測量值的調查(如果適用)應該在開始第一個處理周期前28內完成。在任何研究施用前,在每一周期的第一天,必須重新確定受試者的合格性。下列評估應該在D1的3天內完成(除了周期1,血液檢驗可以在第一天的14天內檢查):1.臨床評估:醫學史、身體檢查、生命指徵(VS),並檢查出血的風險。在其它時候可以按護理的機構標準進行VS評估。2.血液學:全血計數及分類、INR和活化的PTT。3.凝血:如果部分凝血酶原時間(PTT)延長超過正常上限(ULN),需要抽血檢查狼瘡抗凝物(lupusanticoagulant,LAC)。具有延長的aPTT的患者不應該接受VB-111,直至aPTT正常。在檢查狼瘡抗凝物(LAC)陽性的患者中,在重複施用VB-111之前需要陰性測試。對於一直陽性的LAC或抗磷脂抗體(APLA)檢驗,必須在初始陽性檢驗開始的12周內重複所述的檢驗。4.綜合代謝方面:包括電解質、肝功能檢驗(LETS)、血尿氮(BUN)/肌酸酐(Cr)、鈣和鎂。5.尿:收集用於常規分析。6.VB-111特異實驗室:抽血用於:(i)生物學分布:VB-111腺病毒DNA水平和轉基因表達定量。(ii)生物標誌物:將檢驗所有列入本研究的患者的外周血樣品中抗血管生成療法的生物標誌物。分析血漿中循環血管生成性和炎症性生物標誌物:VEGF、PlGF、sVEGFR1、bFGF、IL-1β、IL-6、IL-8、和TNF-α(使用Meso-ScaleDiscovery的多重ELISA平板)以及sVEGFR2和SDF1α(使用R&DSystems試劑盒)。將使用標準流式細胞術計數新鮮樣品中的血液循環細胞。對存檔的組織評估CD31、CD34、VEGFR2和vWF。(iii)腫瘤標誌物:CA-125(iv)抗體:針對AD-5病毒的抗體水平(包括中和抗體)。研究處理輸註:在研究處理輸注前ANC必須≥1,000/mm3並且血小板計數≥100,000/mm3。紫杉醇輸註:抗嘔吐療法:抗嘔吐療法可包括地塞米松10mgi.v.prn、氯羥安定(Ativan)0.5-2.0mgi.v.、和/或康帕嗪10mgp.o.或羥哌氯丙嗪4mgp.o.,基於學院標準。施用順序是紫杉醇然後VB-111。經過具有不大於0.22微米的微孔膜的管路過濾器,以起始劑量40–80mg/m2每周施用紫杉醇,用時超過60分鐘。所有患者在施用紫杉醇之前必須接受術前治療(premedication)以防止嚴重的過敏性反應。患者將口服地塞米松,在處理前一天晚上20mg,在第一次施用處理的早晨20mg,然後如果紫杉醇耐受,則第二周不用口服,只是IV術前治療,劑量逐漸減少,第三周後不再使用。典型的術前治療方案組成如下:在紫杉醇之前30-60分鐘給予10-20mg靜脈內(IV)地塞米松、50mgIV苯海拉明(或其等價物),以及300mg西咪替丁或50mgIV雷尼替丁。根據當地處方,法莫替丁20mgIV可以作為替代物。如果在施用紫杉醇時患者發生了過敏反應,研究人員可以自行決定提高地塞米松劑量。奇數周期的第一天:除了上述操作,將在奇數周期的第一天進行下列程序。腫瘤測量:在周期1的第一天之前(允許長達一個月之前)以及在每個奇數周期(例如周期1、3、5等=每8周)直至病情發展,利用CT進行胸部、腹部和骨盆的腫瘤測量。在施用前、長達施用前3天,進行評估。研究處理輸註:VB-111將在每個奇數周期的第一天施用VB-111直至病情發展。應該在紫杉醇之後施用VB-111。這是基於以下考慮,作為安全防範,研究性藥劑應該在最後施用。沒有預期兩種藥劑會有順序依賴的藥理學改變。雖然預料這會在紫杉醇之後立即施用(1小時之內)的情況下發生,如果有臨床指示,可以晚些施用(24小時內)。具有延長的aPTT的患者不應該接受VB-111直至aPTT正常。LAC和/或APLA檢驗陽性的患者,需要在重複施用VB-111之前進行陰性檢驗。如果檢驗仍然陽性,則不應該進一步施用VB-111。對於一直陽性的LAC或APLA檢驗,從最初檢驗陽性起12周內必須重複檢驗。退熱治療:施用VB-111之前應該施用1000mg的醋氨酚,VB-111之後施用PRN醋氨酚。對於發展了3級發熱的患者或者根據研究人員的自行判斷,可以在接下來施用VB-111之前10分鐘i.v.施用10mg地塞米松。VB-111的準備和輸註:完成紫杉醇輸注後,將對正在禁食或只進食了清淡飲食的患者施用單劑量VB-1113x1012VPs(劑量水平1-2)或1x1013VPs(劑量水平3)。請注意:如果患者具有明顯的發冷,而不是通常的低級和自限性發熱,則建議禁食以避免嘔吐。這不是絕對要求。施用的最終溶液應該在製備後不超過60分鐘內施用。施用後0.5小時允許正常飲食。必須以下列速度施用稀釋的VB-111單一靜脈內輸註:1.對於劑量3x1012VPs:1mL/分鐘2.對於劑量1x1013VPs:最初的10mL為1mL/分鐘,然後剩餘的輸注為3mL/分鐘。VB-111施用後的觀察在第一次施用研究藥物後的最初8小時,研究參與者應該在門診部或輸注區進行觀察,接下來根據臨床指示而定。VB-111分布實驗在下列時間點收集血液和尿液樣品用於VB-111腺病毒DNA表達水平定量:血液:1.0(施用前)2.輸注結束時3.3±0.5小時4.6±0.5小時生命指徵:記錄生命指徵(收縮壓和舒張壓、外周心率、體溫、呼吸率):1.0分鐘(就在施用前)(-15分鐘的範圍)2.開始施用後30分鐘(+/-15分鐘的範圍)3.開始施用後60分鐘(+/-15分鐘的範圍)4.開始施用後4小時(+/-15分鐘的範圍)5.開始施用後6小時(+/-15分鐘的範圍)6.在任何不良事件發生的時候每個周期的第8、15和22天每名患者被要求在禁食狀態下回到門診部,進行下列評估:1.生命指徵:收縮壓和舒張壓、外周心率、體溫、呼吸率。2.血液學:全血計數並分類、INR和活化的PTT。3.凝血:如果需要,跟蹤異常PTT水平。4.尿液:收集用於常規分析血液實驗將在周期1-6的第8天抽血。抽血用於:1.生物學分布:VB-111腺病毒DNA水平以及轉基因表達定量(VB-111輸注後的每個第二周期)2.生物標誌物:分析血漿中循環性血管生成性和炎症性生物標誌物VEGF、PlGF、sVEGFR1、bFGF、IL-1β、IL-6、IL-8、和TNF-α(使用Meso-ScaleDiscovery的多重ELISA平板)以及sVEGFR2和SDF1α(使用R&DSystems試劑盒)。將使用標準流式細胞術計數新鮮樣品中的血液循環細胞。對存檔的組織評價CD31、CD34、VEGFR2和vWF。3.腫瘤標誌物:CA-1254.抗體:針對病毒的抗體水平(包括中和抗體)注意:綜合代謝方面包括電解質、LETS、BUN/Cr、鈣和鎂分析只需要在第一天進行,以及只需要在第1、8、15、22天進行LFTs。如上詳述施用紫杉醇。ANC必須>1,000/mm3並且血小板計數>100,000/mm3抗嘔吐療法:抗嘔吐療法包括凱特瑞(Kytril)750μgi.v.或1mgp.o或樞復寧(Zofran)10mgi.v.或8mgp.o.,和地塞米松10mgi.v.,以及prn氯羥安定(Ativan)0.5-2.0mgi.v.,和/或甲哌氯丙嗪(Compazine)10mgp.o.或奮乃靜(Perphenazine)4mgp.o.,遵照學院標準。研究完成回訪受試者將按上述周期規劃給予組合療法(紫杉醇和VB-111)直至疾病發展。最後一次施用VB-111後,要求每名患者回到門診進行最後的跟蹤訪問,進行相同的臨床評估和常規實驗室樣品採集,對於臨床重要事件,其將被記錄作為未規劃的實驗室評估。不良事件以及相關治療應以在前的研究相同的方式記錄。治療醫師(MD或NP)將對受試者進行體檢。將進行ECG。將對沒能繼續研究或發生疾病發展的患者,進行臨床跟蹤或電話聯繫以評估存活。但是疾病發展後,描述CA125和RECIST的數據將被保存在臨床研究文件中。另外的程序不良事件對於具有任何不良事件的患者應進行完全的支持性措施。在施用後發生的所有不良事件將以病歷報告形式(CRF)被記錄,並記錄強度、應用的治療措施、結果和與研究藥物相關性的評價。對相關的不良事件進行跟蹤,直至消退。對不相關的不良事件進行跟蹤,直至消退或研究結束時。常見副作用包括噁心、嘔吐以及食慾減退。患者也可能會遇到便秘、拉肚子和腹瀉。如果發生腹瀉,增加液體攝入非常重要。如果症狀變得嚴重,患者可能需要住院治療和接受靜脈輸液。施用任何研究性產品都涉及一般的副作用風險。因為VB-111是研究性產品,並不知道其所有的在人體中的潛在副作用。VB-111可能會引起下列副作用的全部、一些或不引起這些副作用。副作用是指你在一項研究中可能發生的不良醫學狀況或預先存在的醫學狀況的惡化。除了下列可能的不良反應,不可預見的或不常見的副作用(可能會比較嚴重或威脅生命)可能會在VB-111單獨給予或與其它治療組合時發生。本研究目的之一是研究VB-111可能的副作用。在該研究過程中,研究醫生會密切監測患者,並與患者討論有關風險、不適和不良反應的任何問題。與VB-111相關的危險很可能的(高於50%的機率會發生)·流感樣症狀,比如發熱、肌肉疼痛、疲勞、發冷,·噁心·嘔吐偶爾的(1-10%的機率會發生)·便秘·肝臟產生了異常高水平的酶,意味著你的肝臟功能不正常,並有可能引起疲勞和黃疸(皮膚和眼睛顏色發黃)。儘管這通常比較輕微而且可逆,但是這也可能嚴重或威脅生命。·脾臟大小的增加通常不引起任何症狀,但是如果脾臟破裂,則可能引起胃痛,並可能威脅生命。·骨髓中新血細胞生成的增加,可能導致增加的血細胞數目,但是白血病風險不會增加。·在給予注射位置的過敏反應,其可能引起一些紅腫。·出血·異常延長的凝血檢驗(PTT)和某些類型的抗體的產生(抗磷脂抗體),其可能干擾正常血液凝固。這可能引起血栓(血凝塊)或出血,其可能需要治療,並可能比較嚴重或威脅生命。·你的傷口癒合能力可能會受到影響。這可以導致感染,並可能需要住院治療。·高血壓·尿中過多的蛋白。這通常是無症狀的實驗室發現,但是,如果過多,可能會引起體液滯留,如腿腫。·頭痛·食慾減退和體重減輕(厭食)·血液中鈉水平降低,其可以引起混亂、癲癇、疲勞和低水平意識。·血液中過多的糖分,如果嚴重,則可能需要住院治療以及緊急治療。·出汗增加(多汗症)罕見的(低於1%的機率會發生)·紅細胞數量低,可引起疲勞和呼吸急促。這有可能需要輸血。·血小板數量低,這可能引起出血和淤青。出血可能會很嚴重或威脅生命,並有可能需要輸血。·白細胞數量輕微升高(可能增加感染危險)·嚴重出血·腎功能可能惡化(急性腎衰竭),這可能需要治療,並且可能很嚴重或威脅生命·在具有涉及腦部的腫瘤的患者中,可能產生腦水腫(例如腦轉移)·腹瀉·心血管併發症:心臟病、心絞痛(胸痛)或血凝塊(血栓),其可能堵塞重要器官的血液供應,並引起中風或對其它器官的病變。·充血性心力衰竭-心臟不能正常泵血,其可導致虛弱和疲勞、流體滯留和流體集於肺中,這可能導致呼吸急促。可能很嚴重或威脅生命。與紫杉醇相關的危險很可能的(高於50%的機率會發生)·輕微至嚴重的過敏反應,可能威脅生命·手足麻木和疼痛,有時惡化需要另外的治療,並且在停藥後可能不會消失·脫髮·肌肉和關節疼痛·疲勞經常(10-50%的機率會發生)·噁心和/或嘔吐·腹瀉·口或喉酸痛·頭昏目眩·頭痛·肝病變·如果藥物從注射的靜脈洩漏到其周圍的皮膚中,則有皮膚刺激和腫脹。·味覺改變·先前的放射位點處的皮膚刺激·皮疹偶爾的(1-10%的機率會發生)·結腸炎症,可能會引起你的腸部運動改變·胰腺炎症,可能會引起短時的腹部疼痛·閃爍的燈光或斑點的感覺·腎臟病變·一種叫作甘油三酯的脂肪形式在血液中水平升高(高甘油三酯血症)·心率減慢·心跳不規則罕見(低於1%的機率會發生)·肝衰竭·癲癇·混亂;情緒變化與活檢相關的危險:活檢通常在成像技術的指導下進行。每個操作在活檢前需要獨立的同意書。該危險可能包括:·疼痛和不舒服。疼痛和不舒服的程度會不同,取決於活檢位點的位置。可以和研究醫生討論這些風險。·在活檢位點少量出血。·在活檢位點觸痛。·在活檢位點結疤。·罕見的在活檢位點的感染。非常少見地,活檢引起的併發症可以威脅生命。對於任何介入性操作,可能發生其它由於出血或器官病變導致的潛在的嚴重併發症。這些可能需要進一步的外科幹預。與放射掃描和X-射線相關的危險:可能會應用CT掃描評估該研究中患者的疾病。這些檢驗的頻率是標準治療。從長期來看,經過很多年,由於對癌症的放射學評估和治療導致形成新的癌症的風險非常低。某些類型的藥物或這些藥物與放射治療的組合,可能會進一步輕微增加形成新癌症的風險。在掃描過程中注射到血管中的造影劑的使用存在很小的風險。它可能使腎功能已經降低的人的腎功能惡化。因此,在參與該研究的過程中將密切監測腎功能。如果患者的腎功能發生任何變化,該患者可能會被移出該研究。非常少見地,一些人會對造影劑有過敏反應(比如蕁麻疹和瘙癢)。嚴重的反應(例如,血壓下降、呼吸困難或嚴重的過敏反應和死亡)非常罕見。生殖性風險:該研究使用的藥物可能會影響胎兒。當參與本研究時,患者不應該懷孕,並且不應該給嬰兒餵奶。患者如果懷孕應立即告知醫生。將對男性或女性研究參與者提供有關預防懷孕的諮詢。合併用藥除了在排除標準中列出的藥物,對合併用藥沒有限制。但是,VB-111不應該與其它藥物混合。從基線直到最後一次施用後1個月的跟蹤回訪,所有在研究中給予的合併用藥將被記錄。將記錄常規藥物的產品名稱、說明、劑量、單位、頻率、起始和停止日期。任何持續的合併用藥將如此記錄,不必重複出現,除非在研究結束前記錄停止用藥日期。止吐方案預計噁心應該是輕微的副作用。建議下列代表性的止吐方案:地塞米松4-8mgPO,或蘿拉西泮(Lorazepam)0.5-1.0mg,或丙氯拉嗪(Prochlorperazine)10mgPO,或甲氧氯普胺(Metoclopramide)10-20mg,化療前30分鐘。阿司匹林對於具有高風險動脈血栓栓塞性疾病的受試者,可以開始或持續施用低劑量的阿司匹林(≤325mg/d)。在研究中出現動脈缺血症狀或出血的受試者,應該對停止藥物進行評估。退熱治療VB-111施用通常與在施用後持續24小時的自限性發熱有關,一些病例中,低級發熱可能延長至施用後2-3天。在VB-111施用前1-2小時應該施用1000mg的醋氨酚,VB-111施用後施用PRN醋氨酚。在發展了3級發熱的患者或者根據研究人員的自行判斷,在接下來施用VB-111之前10分鐘,IV施用10mg地塞米松。其它實驗室分析因為臨床重要事件抽取的實驗室樣品,將記錄為未計劃的實驗室評估。對於具有臨床相關的異常實驗室數值時,將跟蹤檢驗,直至該數值回到正常範圍內和/或發現所述異常的充分解釋。所有該類實驗室研究都將在該研究地進行,除了分布評估,其將被送到獨立中心實驗室(IndependentCentralLaboratory)。如果任何這些結果需要證實,如果可能,將在同一醫院實驗室進行重新檢驗。必須為每個醫院實驗室提供認證證書和正常參考範圍。如果從當地實驗室抽取安全性實驗室,應該盡力為這些實驗室獲得實驗室認證證書和正常參考範圍。在篩選訪問和每6個月或早期撤出的訪問中將進行ECG。研究持續時間每個患者將每周施用紫杉醇和每第二個周期施用VB-111。患者將參加該研究直至病情發展,然後只有他們的文件可供CA125和RECIST詳細信息的收集。患者可以決定在任何時間撤出研究。應該仍然聯繫撤出的患者並詢問有關不良反應(AE)。所有AE,不管是否與藥物或疾病相關,都將記錄在患者的記錄和CRF中。基於到目前為止完成的臨床前研究得到的毒性特徵,以及轉基因表達的特異性,認為一年時間足以確定治療後產生的任何長期毒性。應該大約每2個月聯繫已經撤出的或發生病情發展的患者,調查存活率。劑量限制性毒性(只是周期#1,2和3)在研究計劃的第一個周期確定劑量限制性毒性(DLT),在這些周期中產生DLT的所有患者將被移出研究計劃。在周期1中不允許劑量降低。DLT被定義為>4天的IV級中性粒細胞減少症(ANC100.2°F和ANC<500/mm3)、IV級血小板減少症(血小板<25,000/mm3)以及等級≥III的非血液學毒性,不包括噁心和III級嘔吐。如果他們對對症療法有反應,在VB-111施用後24小時內發生的3-4級發熱不應被視為DLT。對於每周的計劃,如果80mg/m2的紫杉醇是臨床可接受的劑量,則沒有計劃升高超過該劑量。劑量調整–紫杉醇在本研究劑量升高階段的第一周期不會進行劑量調整。在與研究PI討論之後,可以允許具有神經病變症狀的患者有「一周休息」。預計允許4周中有3周的施用計劃,或者如果是交替周期,8周中有7周的施用計劃。如果在這些計劃之一中不能保持施用密度,該患者應被移出研究。如果對紫杉醇的計劃進行了劑量調整,則無需因此對VB-111施用進行調整來遵循紫杉醇的周期。由於毒性恢復或社會義務而中斷施用最長達3周的參與者可允許保持在研究中。注意:28天以後,一旦患者劑量降低,則不允許劑量升高,在接下來的所有周期中該患者應該繼續該劑量。應該根據按照CommonTerminologyCriteriaforAdverseEvents(CTCAE-4)分級顯示最大毒性的體系進行劑量調整。除非特別說明,否則針對血液學毒性的通常劑量調整將基於再處理時間72小時內獲得的血液計數。將基於ANC(表12)、血小板計數(表13)和神經病(表14)確定紫杉醇的施用劑量。表12:每周一次給藥前的中性粒細胞減少症表13:每周一次給藥前的血小板減少症表14:感覺神經病劑量降低圖表一旦紫杉醇劑量被降低,其不應該被重新升高。延長的骨髓抑制的管理到28天時,如果血小板或中性粒細胞不能恢復,可以允許另外7天進行血液學恢復。在第35天未完全恢復(ANC<1,000/mm3或血小板<100,000/mm3)的患者將被移出研究計劃。發熱和中性粒細胞減少症的管理對於發熱和中性粒細胞減少症的患者將以通常方式進行管理。患者需要在重新進行療法前從發熱和活躍的感染問題中恢復。沒有用生長因子的患者將在他們的下一周期中加入Neupogen或Neulasta。使用Neupogen5ug/kg/天的患者將在同一計劃內將劑量升高至10ug/kg。另外的毒性和劑量調整在本試驗中不預見其它毒性,但也有可能出現。發生藥物相關的III或IV級非血液學或非黏膜毒性的患者如果在下一周期的7天內沒有消除或改善到I級,其將被移出研究計劃。發生嚴重III或IV級非血液學/非黏膜毒性的患者如果在下一周期開始的一周內消除或改善到I級,患者對治療有反應並且主要研究人員已經複閱了患者的毒性,其可繼續留在試驗中。對於異常肝功能檢驗的劑量降低如下:未發現紫杉醇引起肝毒性;但是,在嚴重肝功能障礙的患者中其消除延遲。因此,應該根據表15對肝功能障礙的患者的紫杉醇劑量進行調整。表15:基於血清天冬氨酸氨基轉移酶(AST)和丙氨酸氨基轉移酶(ALT)水平的紫杉醇施用劑量血清AST,或ALT紫杉醇1.5mg/dl的升高膽紅素的患者不應接受紫杉醇,直至該異常實驗室數值改善到1,000個細胞/mm3、血小板計數>100,000個細胞/mm3、以及非血液學毒性改善到1級(輕微)或消除。患者在開始重新治療前可延遲多至3周。如果需要多於3周進行毒性恢復,他們應該被移出研究。如果毒性:利益權衡是有益於參與者和臨床醫生,那麼在2級疲勞、腹瀉、脫髮和噁心的情況下可以繼續治療。紫杉醇配製、提供、保存和穩定性紫杉醇(NSC#673089)在28天一個周期的第1、8、15和22天IV輸注紫杉醇40-80mg/m2,用時超過1小時。將按最大體表面積(BSA)2.0m2確定藥物給藥劑量,並根據當天的體重計算BSA。配製紫杉醇以6mg/mL非水溶液提供在多種劑量樣品瓶中:其中包含30mg/5mL、100mg/16.7mL或300mg/50mL紫杉醇。除了6mg的紫杉醇外,每mL的無菌無熱源性溶液中含有527mg純化的EL(聚氧乙烯蓖麻油)和49.7%(v/v)無水醇,USP。保存當於20至25℃(68至77°F)下保存時,未打開的紫杉醇藥瓶可以穩定至包裝上指示的日期。避光。製劑紫杉醇在輸注前稀釋。可以將紫杉醇稀釋於注射用0.9%的氯化鈉,USP;5%葡萄糖注射液,USP;5%葡萄糖和0.9%氯化鈉注射液,USP;或者5%葡萄糖Ringer’s注射液,至終濃度0.3至1.2mg/mL。該溶液在室溫(大約25℃/77°F)和室內光線條件下保持物理和化學穩定長達27小時。為了儘量減少患者暴露於增塑劑DEHP(其可能從PVC輸液袋或組件中浸出),稀釋的紫杉醇應該保存於瓶子(玻璃、聚丙烯)中或塑料(聚丙烯、聚烯烴)中,並通過聚乙烯內襯的施用組件施用。施用將通過具有不大於0.22微米的微孔膜的管路過濾器(inlinefilter)施用紫杉醇。過濾器比如或(結合短進口和出口PVC塗層的管道)的使用不會引起DEHP的明顯浸出。術前治療所有患者將在第一次紫杉醇施用之前使用糖皮質激素、苯海拉明和H2拮抗劑進行術前治療(premedication)以防止嚴重的過敏性反應。對藥物發生嚴重過敏反應的患者可能需要重複術前治療和用稀溶液重複刺激以及慢速輸注。對於紫杉醇嚴重過敏性反應無需進行重複刺激。當對藥物很好耐受後或建立了對樟腦的耐受,口服地塞米松可逐步取消。多西紫杉醇不能被替代。不良反應:有關最新和完整的信息,查閱包裝內說明書。供應者:可從Bristol-MyersSquibbOncology以及常規生產商商售獲得。查閱AmericanHospitalFormularyServiceDrugInformationguide、FactsandComparisons或包裝內說明書獲得更多信息。VB-111說明研究藥物將以每受試者劑量提供在標註的1.8ml冷凍管(聚丙烯)中。每一冷凍管含有1.1ml(1012vp/ml)的體積。配製VB-112被配製為無菌載體溶液。溶液以冷凍形式(低於65℃)提供在一次性使用的塑料旋口瓶中。每瓶含有病毒滴度為1012vp/ml的1.1mL載體和媒介物(10%甘油的磷酸緩衝液)。融化載體溶液,並在稀釋和操作過程中,將載體溶液保持在2-8℃。穩定性和保存VB-111的穩定性研究仍在進行中,到目前為止認為在65℃以下保質期為30個月。打開和/或稀釋的藥瓶不應被重複使用。VB-111藥瓶應在封閉、冷凍(低於65℃)、避光下保存。製劑將如表16所示準備VB-111。表16.VB-111製劑*配藥時可以使用無菌空袋並分別向袋中加入40mlNS+10mlVB-111,或者配藥時可以使用50ml的NS袋,去除多出的體積然後加入VB-111。任一方式都是配藥實踐中可接受的。整個藥物配製過程應在BSCII型室中室溫下進行。融化後,在準備過程中,藥物可在冰水中保持長達3小時。藥物稀釋於室溫鹽水中。在儘可能短的時間內完成藥物的準備和藥物注射,不要超過1小時。準備藥物的藥劑師應該驗證容器上的信息適宜該研究和受試者:產品名稱、濃度、批號。在無菌塑料管中放置需要體積的鹽水(至室溫)。在戴著手套的手之間摩擦融化VB-111溶液藥瓶。立即從每個意欲用於特定受試者的冷凍藥瓶中取出1mlVB-111。每個1mlVB-111都使用新的注射器。將VB-111加入含有事先準備的鹽水溶液的塑料管中。用手渦旋內容物來混合稀釋的藥品。根據患者的重量確定將要使用的體積,抽出注射所需的體積到用於施用的注射器中(參見表16)。準備完成之後進行核對過程:檢查使用的原料藥瓶的正確編號,以及管中剩餘的體積大約如預期一致,並完成藥物計量日誌。藥物溶液準備後,根據藥學程序清潔藥房中的藥物配製區。輸注1.應該根據下列指示通過注射器泵進行稀釋的VB-111單次靜脈內輸注施用:a.劑量水平1-2:應該以1ml/分鐘的速率施用3x1012VPs(15或10.5ml,取決於受試者重量)b.劑量水平3:1x1013VPs(50或35ml,取決於受試者重量)i.以下列速率進行輸註:1.在第一個10分鐘為1ml/分鐘2.剩餘的輸注體積為3ml/分鐘完成施用後30分鐘可為受試者提供常規的飲食。受試者中止滿足下列標準的受試者應該中止研究治療:1.3級神經病持續>7天2.4級高血壓或藥物不能控制的3級高血壓3.腎病症候群4.等級≥2的肺或CNS出血;任何4級出血5.任何等級的動脈血栓栓塞事件6.4級充血性心臟衰竭7.胃腸道穿孔8.任何等級的瘻9.任何等級的腸梗阻10.需要藥物或手術幹預的傷口裂開11.受試者不願意或不能遵守研究要求12.研究者確定受試者繼續療法不再安全13.研究者認為的與研究治療相關的所有4級事件禁忌症在具有已知的先前嚴重的(NCICTC3級/4級)對在EL(聚氧乙烯蓖麻油)中配製的藥物有過敏反應史的患者中禁忌研究藥物治療。臨床檢驗和程序根據表17,在整個研究過程中,以指定的間隔進行一系列的臨床檢驗和操作。表17:臨床檢驗和操作*在VB-111施用日(每兩個周期)在施用前、輸注結束、施用後6小時,以及那個周期(奇數周期)的第8天收集樣品。^施用前收集樣品#將在VB-111施用當天在施用前和施用後30分鐘、60分鐘、4小時和6小時監測生命指徵a:所有體檢、血液檢驗和尿分析必須在登記前14天內進行。腫瘤的放射評估應該在登記前4周內進行。b:在所有接下來的周期的第1、8、15和22天的3天內抽血。CBC分類:CBC並分類和血小板;綜合代謝包括電解質、LFT、BUN/Cr、鈣和鎂。在第8、15和22天只要求LFT和CBC分類。c:預計腫瘤應答將被評估2個周期。d:如果出現新的或增加的蛋白尿,需要24小時尿液。+2浸尿棒需要24小時收集,但是+3浸尿棒需要保持研究藥物並24小時收集。因為蛋白尿不是VB-111施用的特徵,尿分析將在第1、8、15、22天獲得。e:可以在每個周期的第一天的三天內抽血檢查PT、PTT。在PTT延長超過ULN的情況下,應該抽血檢查狼瘡抗凝物(LAC)和抗磷脂抗體(針對β2-GP-1和抗心磷脂的IgG和IgM)。f:可根據學院標準進行腫瘤評估,比如腫瘤應答評估:CT,MRI等。注意:可以計劃以規則的間隔進行腫瘤評估(每8周),這通常與每2個周期一致,但是以固定的間隔以便比較徹底的評估PFS、原發終點。g:在周期1的第1天施用前和周期1的第8、15、22天、周期2的第1天、周期3的第1天施用前和每2個周期直至病情進展,收集特異的生物標誌物。h:組織探查不是本研究的必須部分,但是可選的,有預算涵蓋非臨床指示的活檢或穿刺獲得樣品。完成其沒有特定的時間。參見下面組織收集部分。i.研究完成數據應該在研究治療最後施用起30天的一周內完成。存活數據可以電話收集。效力和安全性評估RECIST標準使用ECIST1.1標準在基線和之後每兩個周期(即周期2、4、6等之後)進行腫瘤應答評估。如果患者持續使用紫杉醇,將繼續正式的評估。使用TumorMetrics進行獨立的成像評估。腫瘤測量可測量的疾病:存在至少一種可測量的病灶。如果可測量的疾病限於單獨病灶,應該通過細胞學/組織學證實其腫瘤性。可測量病灶至少可以在一個維度準確測量的最大直徑>2.0cm的病灶。使用螺旋CT掃描,病灶必須在至少一個維度上>1.0cm,並且對於淋巴結,最小直徑必須>1.5cm。不可測量的病灶所有其它病灶,包括小的病灶(使用常規技術最大直徑<2.0cm或使用螺旋CT掃描<1.0cm),以及其它不可測量的病灶。其包括:骨病變;軟腦膜疾病;腹水;胸腔/心包積液;炎性乳腺疾病;皮膚淋巴管炎/肺炎;未證實的、接著進行成像技術的腹部腫塊,和囊性病變。使用尺子或卡尺,以公制表示法記錄所有的測量值。在儘可能接近治療起始時進行所有基線評估,絕不能超過開始治療前4周。在基線和跟蹤過程中應該使用相同的評估方法和相同的技術來鑑定每個確定的和報告的病變。只有當臨床病變位於淺表時(例如皮膚結節、可觸知的淋巴結)才能被認為是可測量的。對於皮膚病變的情況,建議使用包括使用尺子估計病變大小的彩色攝影術記錄。目標和非目標病變的基線記錄代表每個所涉及的器官最多5個病變的所有可測量病變應該確定為目標病變,並在基線記錄和測量。應該基於病變的大小(具有較大直徑的病變)以及可準確反覆測量的適宜性(通過成像技術或臨床上)選擇目標病變。將計算所有目標病變的最大直徑(LD)的總和並報告為基線總計LD。該基線總計LD將被用作參考,進一步鑑定疾病的可測量維度的目的腫瘤應答。所有其它病變(或疾病位點)應該被確定為非目標病變,也應該在基線記錄。無需測量值,這些病變應該跟蹤為「有」或「無」。應答標準目標病變的評估:完全應答(CR)-所有目標病變消失。部分應答(PR)-以基線總計LD作為參考,目標病變的總計LD至少減少30%。進展(PD)-以從開始治療或一個或多個新病變出現開始記錄的最小總計LD作為參考,目標病變的總計LD至少增加20%。穩定病情(SD)-以開始治療以來最小的總計LD作為參考,既沒有充分的縮減而符合PR,也沒有足夠的增加而符合PD。非目標病變的評估:完全應答(CR)-所有非目標病變消失,並且腫瘤標誌物水平正常。不完全應答(非-CR)/非進展(非-PD)-一直存在一個或多個非目標病變或/和維持腫瘤標誌物水平在正常限定值之上。病情發展(PD)-出現一個或多個新的病變。存在的非目標病變的明確發展。儘管僅有明確的非目標病變的發展屬例外,但是在這種情況下,治療醫生的意見起決定作用,之後複閱小組(或研究的首席/主要研究者)應該對該進展狀態進行確認。評估最佳總體應答最佳總體應答是指從開始治療直至病情進展/復發以來記錄的最好的應答(對於進展性疾病,以自治療開始記錄的最小的測量值作為參考)。通常,患者的最好應答評估將取決於測量和證實標準的完成,如表18所示。表18:基於目標、非目標和新病變的患者總體應答目標病變非目標病變新病變總體應答CRCR無CRCR非CR/非-PD無PRPR非-PD無PRSD非-PD無SDPD任何有或無PD任何PD有或無PD任何任何有PD健康狀況全面惡化需要中止治療、而當時沒有病情進展的客觀證據的患者將報告為」臨床惡化」。甚至在中止治療後也要盡一切努力記錄其客觀進展。在一些情況下,區分殘留病症和正常組織可能會比較困難。當完全應答的評估依賴於這種判斷時,建議在確認完全應答狀態前進行殘留病變檢查(微針吸取/活檢)。確認:若指定為部分應答(PR)或完全應答(CR)的狀態,腫瘤測量值的變化必須在首次符合應答標準後經過進行不少於4周的反覆研究來確認。在SD的情形下,在以6-8周的最小間隔進入研究後,跟蹤測量值必須滿足SD標準至少一次。Rustin標準:如果只有CA125是可評估的(升高超過35U/ml),應根據GCIG而不是Rustin標準定義應答。GCIGCA125應答定義:與治療前樣品相比,如果CA125水平在有至少50%的降低,則產生了CA125應答。該應答必須被確認並持續至少28天。只有當患者的治療前樣品至少是正常上限的兩倍,並且在開始治療的2周內,他們才可以根據CA125進行評估。如果CA125有確認的升高(兩次記錄)或者之前的正常CA125升高至≥2xULN(兩次記錄),則發生了基於CA125的進展。預期的不良事件在臨床前小鼠毒理學研究中,VB-111引起最小的毒性。觀察到輕微貧血、輕度的血小板減少症、輕度的白細胞增多、脾腫大和骨髓增生。也觀察到與臨床病理學無關的短暫的肝酶升高。全身性施用腺病毒載體耐受較好。流感樣症狀(發熱、疲勞、發冷、噁心和/或嘔吐)是最常見的不良事件。在參與I/II期試驗的幾個患者中觀察到無症狀的aPTT延長和陽性LAC。另外,在II期患者中報告了唯一一例嚴重腹瀉。大部分靜脈內注射的腺病毒顆粒集中於肝臟,其因此引起了以急性轉氨酶和血管損傷為特徵的炎症反應。抗血管生成劑的主要不良反應為傷口癒合障礙、出血、血栓栓塞和心血管疾病、高血壓和蛋白尿。相關研究分布根據附錄I收集所有患者的血液樣品進行分布評估。在最大耐受劑量組或最高劑量組檢查這些樣品的腺病毒和進行VB-111轉基因水平定量。在預先確定的施用後時間點分別通過血液的Q-PCR和Q-RT-PCR測定血液中病毒DNA水平和轉基因水平,從而進行分布評估。將在所有預定的時間點收集所有患者的樣品。對來自開始於最高劑量的患者的樣品進行檢查,並繼續較低劑量的檢查。對施用後未發現可檢測水平的病毒DNA的樣品將不進行轉基因水平的檢查,並且不進行之後時間點的評估。抗體將收集血清樣品用於分析針對腺病毒的抗體水平(總IgG和中和抗體)。血管生成的生物標誌物目前沒有針對抗血管生成療法的生物標誌物,但是已經確定了幾種候選的生物標誌物[Jain2009]。將在列入本研究的所有患者外周血液樣品中對此進行檢查。進行血漿分析以評估循環血管生成性和炎症性標誌物:VEGF、PlGF、sVEGFR1、bFGF、IL-1β、IL-6、IL-8、和TNF-α(使用Meso-ScaleDiscovery的多重ELISA平板)以及sVEGFR2和SDF1α(使用R&DSystems試劑盒),其中使用已經確定的方法,樣品一式兩份進行檢查[HorowitzClinicalOvarianCancer2011付印中]。將使用標準流式細胞術計數新鮮樣品中的血液循環細胞。定量分析終點是治療後血液單個核細胞中循環性CD34brightCD45dimCPC或VEGFR2+CD45+單核細胞級分發生變化,如之前所述的[HorowitzClinicalOvarianCancer2011列印中]。將對歸檔組織評價CD31、CD34、VEGFR2和vWF。組織收集組織探查不是本研究的必須部分,但是可選的,若有預算支持非臨床指示的活檢或穿刺獲得樣品。完成其沒有特定的時間。VB-111首次施用後的一到兩個月被認為是理想的時間。預計20-50%的參與者可能會有合適的區域用於安全活檢或適合腹水穿刺。統計方法的說明將使用標準的「3+3」設計確定VB-111的MTD。也就是,起始將3名患者以特定的劑量水平進行治療。如果多於一名患者發生DLT,則停止升高,下一個較低的劑量水平被接受為MTD。如果沒有DLT發生,則開始升高至下一個較高劑量水平。如果發生一例DLT,另外3名患者將被加入到那個劑量水平。如果這3名患者沒人發生DLT,將升高VB-111至下一個較高的水平,但是如果一名或多名患者發生此類事件,則將停止升高,並將MTD定義為該下一個較低的劑量水平。利用這一升高方案,表19給出了對於多種假想的潛在毒性率升高至下一個更高劑量水平的可能性。表19:基於DLTVB-111劑量升高的可能性從表19可以看出,如果發生DLT的潛在風險71%的機率升高該組合,如果潛在風險91%的機率升高。相反,如果發生DLT風險>50%,則最高有17%的機率升高,而如果風險>60%,則有<8%的機率。客觀應答的評估將主要是描述性的。通常來講,將計算總體應答率和相應的95%置信區間。II期方面的設計將評估由RECIST標準或CA125(GCIG,不是Rustin標準)確定的應答率,以及描述安全性特徵並表徵不良事件及毒性。根據報告的復發卵巢癌患者對化療劑-抗血管生成劑組合的應答,選擇30%作為目標應答,通常在20-25%範圍內。設計為兩步驟(two-stage)優化設計,其中最初的10名患者在步驟1被列入。這將包括在I期開始紫杉醇劑量為80mg/m2的參與者(即劑量水平2和3的參與者)。從本研究I期部分的劑量水平2和3組列入2到6名患者,在中期效力分析之前另外8到4名參與者將被列入到II期第一個步驟。如果觀察到一個應答或沒有應答,那麼該試驗將停止招募。否則,如果有兩名或多名應答,那麼另外19名參與者將被列入(即可能最多共29名)到II期中(那麼對於整個研究總共將有I期的2-18名與II期的0-29名重疊)。如果有5名或更少的應答,那麼該療法沒有進一步研究的必要。如果真實RR≤10%,在步驟1結束該試驗的機率至少是74%。如果真實RR≥30%,那麼該試驗在步驟1停止的機率≤15%。最終分析的能力是:80%拒絕H0:RR≤10%利於H1:RR≥30%,目標類型-1,錯誤率5%。如在步驟I發現超過2名發生IV級的GI穿孔將中止該試驗。在任何時間如果有3名GI穿孔,那麼該試驗將被中止。假設真實的GI穿孔率是4%,那麼在步驟I觀察到超過2次事件的可能性是6.2%,從整個試驗觀察到3次以上事件的可能性是2.7%。定量的和半定量的數據比如IHC數據將被視為初步試驗數據,使用描述性統計學和非參數分析,試圖探討相關性,承認這些樣品的數量不足以得出明確的結論,但是在多項研究中使用同一組分析的優勢,使我們能夠評價對效力和毒性潛在地最有用的預測指標。生物標誌物將主要使用非參數方法(例如Wilcoxon符號秩或秩和檢驗)評價VB-111對CEC的影響、相關性/預見性措施,所有統計學顯著性檢驗都是雙側的,並且對多重比較不做調整。顯著性水平計算(雙側)95%的置信水平下的置信區間。實驗室檢查血管生成生物標誌物分析(當地)進行血漿分析以評估循環血管生成性和炎症性生物標誌物VEGF、PlGF、sVEGFR1、bFGF、IL-1β、IL-6、IL-8、和TNF-α(使用Meso-ScaleDiscovery的多重ELISA平板)以及sVEGFR2和SDF1α(使用R&DSystems試劑盒)。使用標準流式細胞術計數新鮮樣品中的血液循環細胞。將對存檔組織評價CD31、CD34、VEGFR2和vWF。腫瘤標誌物:CA-125。在室溫EDTA管中的每個時間點10cc總血漿將被分析。將在下列時間點收集樣品:1.在基線2.周期1:研究藥物施用前第1天,第8、15和22天3.周期2:研究藥物施用前第1天4.周期3開始後的每2個周期,研究藥物施用前,直至病情發展。抗體檢查針對Ad-5病毒的抗體(包括IgG和中和抗體)的效價將被收集用於分析。在下列時間點收集樣品:1.在每一周期:第1、8、15和22天在研究藥物施用前;2.在研究完成回訪時。以下列方式收集並準備血液:1.將6ml血液收集到沒有抗凝劑的管中2.將樣品在室溫放置1小時,然後在2-8℃保存過夜以進行血塊凝縮。3.在第二天離心血液。4.將提取大約2ml的血清,分裝成4管(每管0.5ml,在1.5ml的Nalgene100中,總計2ml),保存於-20℃直至運輸去分析。生物學分布檢查進行下列檢查:1.血液生物學分布:病毒DNA和轉基因表達2.尿液生物學分布:病毒DNA在下列時間點收集全血樣品用於生物學分布檢查:1.在每個奇數周期的第1天和第8天在研究藥物施用前。2.在VB-111施用日:a.輸注前(與第1天樣品相同)b.輸注結束時c.3±0.5小時d.6±0.5小時e.在研究完成回訪時以下列方式準備血液樣品:1.收集每一時間點的血液到4個含有EDTA的管中(0.75ml/管)a.兩管用於病毒DNA分析b.兩管用於轉基因表達分析2.管子應該標記受試者號碼、名字首字母、日期和樣品收集時間,並保存於-70℃或低於-70℃的冰箱中。按下列方式收集尿液:1.從保存的總收集體積中收集兩份1-2ml的尿液樣品。2.尿液樣品和總收集體積將冷凍保存直至進一步分析。結果對6名患者施用VB-111,每名患者在研究開始前都具有持續至少一年的輸卵管或卵巢上皮癌,劑量為每名受試者3x1012VPs與紫杉醇(40mg或80mg)的組合,如圖3所示。患者沒有表現任何與VB-111相關的嚴重不良事件。沒有觀察到劑量限制性毒性。