一種鐵基磁性納米晶磁共振T1造影劑的製備方法及其應用與流程
2023-08-08 06:19:51 1
本發明涉及一種超小鐵基磁性納米晶磁共振t1造影劑的製備方法以及其在磁共振t1加權成像中的應用。
背景技術:
近年來,隨著納米醫學的發展,納米材料在藥物傳輸、疾病診斷以及治療等生物醫學領域展現出廣闊的應用前景。其中,超順磁性納米顆粒作為造影劑在磁共振成像方面越來越成為研究熱點。磁共振成像作為一種先進的醫學影像診斷工具,可以實時無創地提供高空間解析度的軟組織解剖學信息,對疾病進行診斷與研究,同時可用於腫瘤的早期診斷。造影劑通過提高造影區域與背景的對比度可以顯著提高磁共振成像的靈敏度。根據質子的縱向弛豫與橫向弛豫效應造影劑可以分為兩大類:可加速縱向弛豫速率的是t1造影劑,可加速橫向弛豫速率的是t2造影劑。在磁共振成像中,發生加速縱向弛豫的區域會變亮而發生加速橫向弛豫的區域會變暗。最初,超順磁氧化鐵主要作為t2加權造影劑,例如商業化的菲力磁(feridex)就是很典型的t2造影劑。但是在臨床使用中,t2加權造影劑有一定的局限性。首先,其變暗的效果與組織內部出血或鈣化、金屬沉積等低信號區域易混淆導致誤診;而且,t2造影劑的高磁矩會誘導局部磁場的擾動,引起所謂的「bloomingeffect」,這個效應會放大影像部位而使得暗的圖像模糊。因此相比t2造影劑,t1造影劑更有可能獲得精準的高分辨圖像。目前臨床廣泛使用的t1造影劑主要是釓類配合物,這些配合物在臨床使用過程中存在諸多不足。1,由於快速的腎代謝,導致體內循環時間短,難以滿足需要長掃描時間窗的高分辨成像;2,釓類配合物分解出自由的釓離子毒性大,而且美國食品藥物監督管理局(fda)已經警告致命的腎源性系統纖維化與釓配合物造影劑有很大的關聯。為了解決上述的釓配合物t1造影劑和傳統超順磁氧化鐵t2造影劑在磁共振成像中存在的問題,開發新型的高效低毒磁共振鐵基t1造影劑迫在眉睫。
技術實現要素:
本發明的目的在於提供一種超小鐵基磁性納米晶磁共振t1造影劑的製備方法以及其在磁共振t1加權成像中的應用。
本發明研究發現超小鐵氧體納米晶可以作為高效的t1造影劑。根據理論研究表明,弛豫增強由外球與內球機制共同起作用而決定。t1造影劑的性能主要有兩個因素決定,縱向弛豫效能r1和橫向弛豫效能與縱向弛豫效能的比值r2/r1,r1越大且r2/r1越小t1造影劑性能越好。根據內球機制,超小鐵基磁性納米晶具有高的比表面積可以增大與水分子的相互作用,同時mn、ni等摻雜可以減小水分子在納米顆粒表面相互作用的停留時間,可以有效提高縱向弛豫效能r1。根據外球機制,超小尺寸(小於5納米)的納米晶具有小的磁化強度值從而限制了其t2弛豫效應,從而降低了r2/r1。目前鐵基磁性納米顆粒主要是通過高溫熱分解法,共沉澱法,反膠束法和溶膠凝膠法製備,因為只有高溫熱分解法可以實現成核與生長分離以及擴散限制生長,因此,通過高溫熱分解法製備的鐵基磁性納米顆粒單分散性最好。就目前所報導的文獻,通過高溫熱分解所製備的鐵基磁性納米顆粒尺寸相對比較大(大於5納米),通常被作為t2造影劑。本發明通過使用芥酸鐵,油酸錳/鈷/鎳/鋅作為前驅物,製備出超小(小於5納米)的鐵基磁性納米晶,而且本發明所製備的超小鐵基磁性納米晶可以作為新型t1造影劑。
本發明的具體實現過程如下:
一種鐵基磁性納米晶mfe2o4的製備方法,包括如下步驟:
(1)將三價鐵鹽與芥酸反應生成前驅物1芥酸鐵;
(2)將二價金屬鹽與油酸反應生成前驅物2,所述二價金屬離子為fe2+、mn2+、ni2+、co2+或zn2+;
(3)將前驅物1、前驅物2和表面活性劑溶入高沸點有機溶劑高溫熱分解製得鐵基磁性納米晶mfe2o4,其中m為fe2+、mn2+、ni2+、co2+或zn2+。
在上述方法中,使用芥酸鐵作為前驅物,發明人發現使用該前驅物可以製備出超小鐵基磁性納米晶。
上述步驟(3)中,所述表面活性劑選自油醇、油酸、油胺、10-22個碳原子的醇或胺、三正辛基氧化膦(topo)、三正辛基膦(top)、油酸鈉等。所述的高沸點有機溶劑為二苯醚(沸點257℃)、十六烯(沸點274℃)、二辛醚(沸點287℃)、二苄醚(沸點298℃)、十八烯(317℃)、二十烯(沸點330℃)、三辛胺(沸點365℃)。
上述步驟(3)中,所述表面活性劑與前驅物(1和2之和)的摩爾比為1:50-50:1。
上述步驟(3)中,在惰性氣體保護下將前驅物1、前驅物2、表面活性劑、高沸點有機溶劑混合均勻,混合物先加熱到80-200℃保持10-120分鐘,然後升溫至200-350℃回流反應10-120分鐘,反應結束冷卻至室溫,加入乙醇/甲醇/丙酮得到沉澱,沉澱經過離心洗滌後重新分散在氯仿/正己烷/甲苯中製備形成磁流體。
前驅物在溶劑中的摩爾濃度為0.001-100mmol/ml。
將前驅物1、前驅物2和表面活性劑溶入高沸點有機溶劑高溫熱分解反應,升溫速率為1-20℃/min。
將前驅物1、前驅物2和表面活性劑溶入高沸點有機溶劑高溫熱分解反應,優選的反應溫度為180-350℃。
由於上述方法製備的超小鐵基磁性納米晶具備磁共振t1增強特性,超小鐵基磁性納米晶應用於造影劑增強的磁共振t1加權成像,特別是將超小鐵基磁性納米晶進行表面修飾與靶向性修飾後用於活體磁共振成像,具體方法為:用水相分散的配體對製得的超小鐵基磁性納米晶進行表面修飾與靶向性修飾後進行活體磁共振成像。
表面修飾所用的配體選自聚乙二醇類衍生物、脂質體類衍生物、多巴胺、3,4-二羥基苯基丙酸(3,4-dihydroxyhydrocinnamicacid)等生物相容性優良的配體,靶向性修飾使用分子、rna、抗體等可靶向腫瘤的物質。
作為優選方案,所製備的超小鐵基磁性納米晶進行膜過濾、超濾離心、透析等處理。
本發明的優點和積極效果:本發明所提供的超小鐵基磁性納米晶的製備方法操作簡單、製備成本低。超小鐵基磁性納米晶產物具有純度高、粒徑小且粒徑分布窄、尺寸可控、低生物毒性等優點,可以作為磁共振成像t1造影劑,具有廣泛的應用前景。
附圖說明
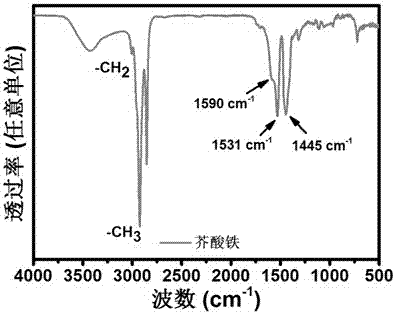
圖1是製備的芥酸鐵紅外光譜圖(ftir);
圖2是製備的芥酸鐵的核磁共振氫譜圖1hnmr;
圖3是製備的芥酸鐵質譜圖;
圖4(a)和(d)是本發明實施例1所製備的2nmmnfe2o4納米晶的透射電鏡(tem)圖,圖4(b)和(e)是本發明實施例2所製備的3nmmnfe2o4納米晶的tem圖,圖4(c)和(f)是本發明實施例3所製備的3.9nmmnfe2o4納米晶的tem圖;
圖5本發明實施例中所製備的2nm、3nm和3.9nmmnfe2o4納米晶的x射線粉末衍射(xrd)圖;
圖6(a)本發明實施例中所製備的2nm、3nm和3.9nmmnfe2o4納米晶的磁化強度隨外加磁場變化圖(磁滯回線);圖6(b)本發明實施例中所製備的2nm、3nm和3.9nmmnfe2o4納米晶的磁化強度隨溫度變化圖;
圖7a本發明實施例中所製備的2nm、3nm和3.9nmmnfe2o4納米晶進行mri測試的t1加權信號圖;圖7b本發明實施例1所製備2nm、3nm和3.9nmmnfe2o4納米晶進行mri測試的1/t1-[fe+mn]濃度的曲線示意圖。
具體實施方式
為使發明的目的、技術方案及優點更加清楚明白,以下參照附圖並舉實施例,對發明進一步詳細說明。
實施例1:2nmmnfe2o4納米晶的製備
(1)前驅體的合成
芥酸鐵:5g六水合三氯化鐵和18g芥酸溶於200ml甲醇,把混合溶液充分攪拌後,向其中滴加200ml甲醇含4gnaoh的溶液。生成的沉澱用甲醇和超純水反覆洗劑,放入真空乾燥箱中30℃乾燥4小時。
圖1是所製備的芥酸鐵的紅外光譜圖,從圖中可以看出,所製備的芥酸鐵顯示出羧酸配合物的特徵振動峰,芥酸鐵的特徵振動峰在1590cm-1,1531cm-1和1445cm-1,其中1590cm-1和1531cm-1是芥酸鐵中羧基官能團的非對稱振動峰;1445cm-1是芥酸鐵的中羧基官能團的對稱振動峰。
圖2是所製備的芥酸鐵的核磁共振氫譜圖。將少許芥酸鐵與油酸錳溶解在氘代氯仿中,裝入核磁管進行測試。從圖2測試結果可以看出芥酸鐵不同化學環境下氫原子的化學位移與模擬結果都能匹配。
圖3是所製備的芥酸鐵的質譜圖。將少許芥酸鐵與油酸錳分散在甲醇或乙腈中進行質譜分析。
油酸錳:4g四水合氯化錳和14g油酸溶於150ml甲醇,把混合溶液充分攪拌後,向其中滴加200ml甲醇含4gnaoh的溶液。生成的沉澱用甲醇和超純水反覆洗劑,放入真空乾燥箱中30℃乾燥4小時。
(2)2nmmnfe2o4納米晶的製備
分別稱取3g芥酸鐵、2g油酸錳和0.5g油醇,將其溶解到8g二苄醚中,氮氣氛圍下攪拌充分混合均勻;
混合物以15℃/min的速率升溫至245℃,回流45min;
混合物降溫到室溫,10ml丙酮加到混合物,離心分離沉澱物,將沉澱物分散於10ml甲苯,重複3次。
實施例2:3nmmnfe2o4納米晶的製備
前驅物芥酸鐵和油酸錳的合成參考實施案例1。
3nmmnfe2o4納米晶的製備:
分別稱取4g芥酸鐵、3g油酸錳、0.5g油醇和1g油酸,將其溶解到8g二辛醚中,氮氣氛圍下攪拌充分混合均勻;
混合物以15℃/min的速率升溫至245℃,回流45min;
混合物降溫到室溫,10ml甲醇加到混合物,離心分離沉澱物,將沉澱物分散於10ml正己烷,重複3次。
實施例3:3.9nmmnfe2o4納米晶的製備
前驅物芥酸鐵和油酸錳的合成參考實施案例1。
3.9nmmnfe2o4納米晶的製備:
分別稱取5g芥酸鐵、3g油酸錳、1g油醇和1g油酸,將其溶解到8g十八烯中,氮氣氛圍下攪拌充分混合均勻;
混合物以15℃/min的速率升溫至255℃,回流45min;
混合物降溫到室溫,10ml乙醇加到混合物,離心分離沉澱物,將沉澱物分散於10ml氯仿,重複3次。
將大約2µl所製備的2nm、3nm和3.9nmmnfe2o4納米晶滴在鍍有碳膜的cu網上,自然乾燥後,做透射電鏡。圖4分別是實施案例1、2、3所製備的2nm、3nm和3.9nmmnfe2o4納米晶tem圖。從圖中可以看出,所製備的2nm、3nm和4nmmnfe2o4納米晶粒徑均勻,對納米晶進行尺寸分布統計,結果顯示平均尺寸分別為2nm、3nm和3.9nm。
圖5是實施案例1、2、3所製備的2nm、3nm和3.9nmmnfe2o4納米晶的xrd圖(cu/kα靶,λ=0.15418nm)。從圖2中可以看到反尖晶石結構的mnfe2o4納米晶的(111)、(220)、(311)、(400)、(511)、(440)和(622)衍射峰,根據最強的(311)衍射峰的2θ=34.980°,由謝樂公式計算得到晶粒尺寸分別為2.07nm、2.98nm和4.29nmmnfe2o4納米晶。
圖6a是實施案例1、2、3所製備的2nm、3nm和3.9nmmnfe2o4納米晶的磁化曲線。測試溫度為300k。從圖6a中可以看出,2nm、3nm和3.9nmmnfe2o4納米晶的飽和磁化強度分別為18.59、24.91和29.18emu/g,其矯頑力和剩磁均為0,表明所製備的2nm、3nm和3.9nmmnfe2o4納米晶均為超順磁性。圖6b是實施案例1、2、3所製備的2nm、3nm和3.9nmmnfe2o4納米晶的磁化強度隨溫度變化圖。測試外加磁場為20oe。從圖6b中可以看出,2nm、3nm和3.9nmmnfe2o4納米晶的轉變溫度分別為6、12.5和15k。
在對所製備的2nm、3nm和3.9nmmnfe2o4納米晶進行mri測試之前,要對其進行轉水相:使用表面聚合物為磷酸化聚乙二醇單甲醚(po-mpeg),具體操作步驟見中國專利201110263936.x。
圖7a是對實施案例所製備的2nm、3nm和3.9nmmnfe2o4納米晶轉水相後進行mri測試的t1加權信號圖。從上到下依次為2nm、3nm和3.9nmmnfe2o4納米水溶膠進行mri測試的t1加權信號圖;每一排從左到右依次為濃度逐漸減低的mnfe2o4納米晶水溶膠。從圖7a中可以看出2nm、3nm和3.9nmmnfe2o4納米溶膠均具有t1加權信號,而且,t1加權信號均隨著濃度的增加而增加。
圖7b中分別是本實施例1、2、3所製備的2nm、3nm和3.9nmmnfe2o4納米晶轉水相後進行mri測試的1/t1-[fe+mn]濃度曲線圖。由圖計算得,2nm、3nm和3.9nmmnfe2o4納米溶膠進行mri測試的r1值分別為8.43、8.23和6.98mm-1s-1。
以上所述僅為發明的較佳實施例而已,並不用以限制發明,凡在發明的精神和原則之內,所做的任何修改、等同替換、改進等,均應包含在發明的範圍之內。