二芳基硫醚衍生物、其製備方法以及作為五羥色胺轉運體靶向顯像劑的用途與流程
2023-07-28 21:25:06 2
本發明屬於放射性藥物化學領域,涉及一類二芳基硫醚衍生物、其製備方法以及作為五羥色胺轉運體靶向顯像劑的用途。
背景技術:
五羥色胺轉運體(sert)是一種選擇性五羥色胺(5-ht)跨膜轉運蛋白。sert能夠重新攝取突觸間隙的5-ht,從而調節神經信號的傳導。在中樞神經系統中,存在於神經元突觸前膜的sert,能夠選擇性地將神經遞質5-ht由突觸間隙轉回到細胞內胞液中,從而終止該遞質對突出後膜5-ht受體的作用,因而在5-ht釋放後的再攝取過程中發揮極其重要的作用。研究表明,sert濃度的變化與許多神經精神異常疾病有關,例如抑鬱症、憂鬱症、焦慮症、精神分裂、藥物濫用、酒癮、飲食失調、阿爾茲海默氏症(alzheimer’sdisease)以及帕金森氏症(parkinson’sdisease)等。sert也是普遍使用的抗憂鬱處方藥物的主要作用靶點。
正電子發射斷層計算機掃描(pet)顯像可以無創、動態、定量地研究人體內的化學過程和生理生化過程,在活體水平研究生命物質的代謝,受體的分布和功能,基因調控的變化等,這些使核醫學的發展達到真正意義上的分子水平。pet顯像離不開正電子核素標記的pet藥物,其中18f正電子核素(半衰期為110分鐘)比11c(半衰期為20分鐘)具有相對較長的半衰期,適合商品化生產和運輸,是當前pet顯像藥物的理想放射性核素,18f藥物成為研究的熱點。因此,以非侵入性的pet和單光子斷層掃描(spect)活體顯像技術研究sert在腦部區域的分布,將有助於探討5-ht系統在神經精神性異常疾病的病理生理學及治療上所扮演的角色,認識sert密度的變化與精神紊亂疾病及精神退行性疾病病理的關係,也可為治療藥物作用機制研究、個體化治療策略制定及療效評估等提供非常有效的信息。
過去十多年中以sert為靶點的顯像劑的研究取得了較大的進展,國際上開發出了一系列有潛力的化合物,目前有多個國家在進行該類顯像劑的研發或者臨床前研究,該領域已經成為當今世界在顯像劑研究方面的熱點,主要集中於託烷環類、硝基喹啉哌嗪類、二芳基硫醚類先導化合物的修飾方面。近年來,在二芳基醚類化合物上取得了突破性進展,發展了一批體內外性質優良的分子探針。該類化合物是基於抗抑鬱藥物moxifetin和403u76(其結構式如下)(oyas.etal,5-iodo-2-[[2-2-[(dimethylamino)methyl]phenyl]thio]benzylalcohol,jmedchem,1999;42:333–335)發展而來的,合成簡單,沒有立體異構體的存在,分離純化容易,在體內外具有較高的親和性和選擇性。其中用於spect顯像的[123i]adam(其結構式如下)和用於pet顯像的[11c]dasb(其結構式如下)已經用於人腦sert的臨床顯像研究。但對於18f標記pet顯像劑而言,目前還沒有臨床廣泛應用的顯像劑問世,國內外研究者正加緊研發具有sert靶向特異結合的18f顯像劑。
sert又是抗抑鬱藥物的主要作用靶標,二芳基硫醚類配基與sert具有靶向特異結合,其衍生物作為sert抑制劑,已成為治療抑鬱症和精神神經疾病的藥物。目前新的sert靶向化合物還在繼續研究,其目的是尋找性能更優秀的治療藥物。在sert/pet顯像劑的開發中,本發明人研究發現,當在骨架b環4位引入側鏈(如下結構式所示),能夠調節該系列化合物的藥代動力學性質,因此本發明人提供一種含f的二芳基硫醚類衍生物,在骨架b環4位引入了不同側鏈,提高了與sert的靶向結合性,從而改善目前此類化合物作為sert顯像劑存在的缺陷。同時,當用18f-標記作為放射性靶向藥物研究時,使其具有更高的體內靶/非靶比值,更適宜的體內動力學性質,有望滿足今後臨床sert顯像需要。
技術實現要素:
本發明的目的在於提供一種二芳基硫醚衍生物,該類化合物具有較高的腦內sert(五羥色氨轉運蛋白)特異性結合效果,同時具有較好的體內生物代謝性質,是較好的腦內攝取的靶向結合顯像藥物。
本發明的另一目的在於提供一種上述二芳基硫醚衍生物的製備方法。
本發明的還一目的在於提供上述二芳基硫醚衍生物作為五羥色胺轉運體靶向顯像劑的用途。
根據本發明的一個方面,本發明提供結構式如下的二芳基硫醚衍生物:
其中,r為直接鍵、(n為1-4的自然數)、c1-4的亞烷基、未取代或c1-4烷基取代的-ch=ch-或者-c≡c-;
y為氟原子,可以為[18f]或者[19f]。
根據本發明的另一個方面,本發明提供一種上述二芳基硫醚衍生物的製備方法,該方法包括:在鹼存在下,2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羥基)-苯)硫醚與進行親核取代反應,得到上述結構式所示的二芳基硫醚衍生物,其中所述的鹼為碳酸鉀、氫化鈉或者碳酸銫等。該反應式如下:
根據本發明的又一個方面,本發明的二芳基硫醚衍生物,為含有[19f]或[18f]的化合物,[19f]-二芳基硫醚衍生物表現出對中樞sert的高親合力,同時相對於dat(多巴胺轉運體)以及net(去甲腎上腺素轉運體)對sert的高選擇性。因此,相應的[18f]-二芳基硫醚衍生物,可以作為sert/pet顯像劑,這些化合物有望獲得較高的腦攝取值,合理的藥代動力學性質,有望開發出優良的sert/pet顯像劑。
附圖說明
圖1為化合物a5經過oasishlb柱子初步提純後hplc譜圖中的放射性峰;
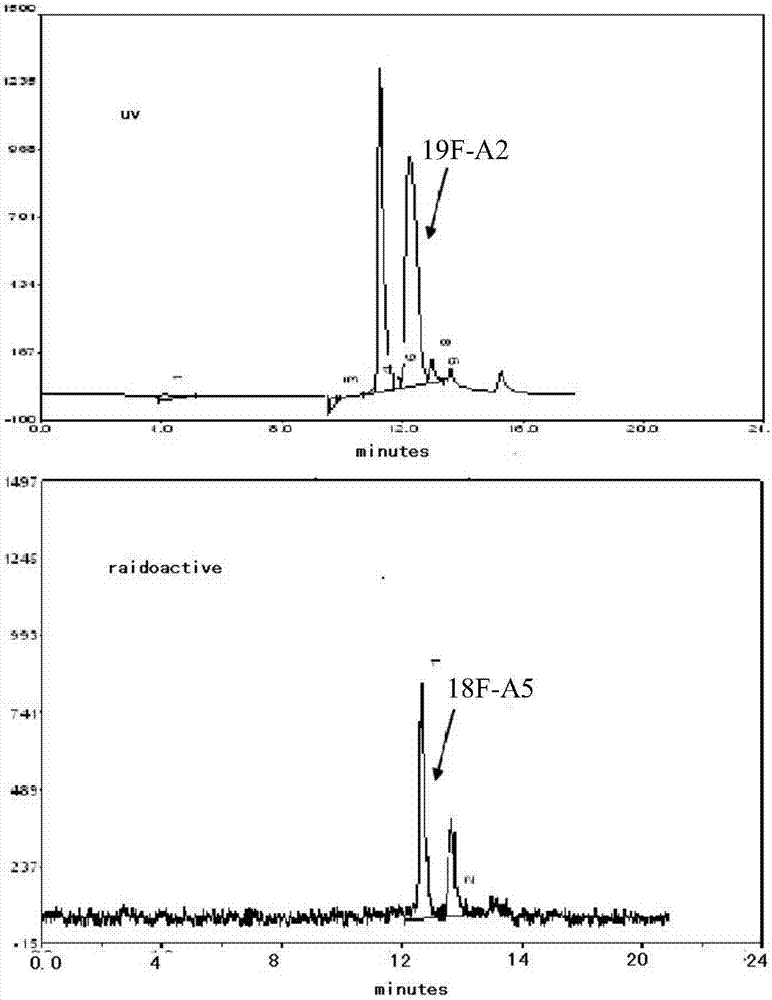
圖2為化合物a5即[18f]-a5的hplc確認,即放射性產物[18f]-a5與化合物a2即[19f]-a2共注射譜圖;
圖3為[18f]-a6的hplc確認,即放射性產物[18f]-a6與[19f]-a3化合物共注射譜圖。
具體實施方式
下面更具體地描述本發明。
在有機化學領域,對於氟化合物,如果沒有說明,一般是指非放射性的[19f]的化合物。因此,在本發明中對於具體的化合物,如果沒有特別指明,則是指非放射性的[19f]的化合物。
在本發明優選的實施方案中,所述二芳基硫醚衍生物,其中,r優選為(n為1-4的自然數)、-ch=ch-或者-c≡c-。
通過以下實施例更具體地說明本發明的二芳基硫醚衍生物及其製備,以及對sert的高親合力,同時相對於dat和net對sert的高選擇性,但要理解的是本發明的範圍不限於這些實施例。
實施例1
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-氟乙氧基)-乙氧基)-苯)硫醚(化合物a1)的合成
合成反應方程式:
合成方法:
將0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)甲氨基-4-羥基)-苯)硫醚(合成參考文獻:(shunichioyaetal,nuclearmedicineandbiology,34(2007),129–139)溶解在4mldmf(hplc純)中,加入2倍摩爾量的碳酸鉀以及1-對甲苯磺醯基-2-氟乙氧基乙烷(1.2倍摩爾量),之後通入氮氣3-5min,將反應液在80-90℃條件下油浴加熱過夜。反應完成後加入30ml水終止反應,並用乙酸乙酯萃取產物,合併有機相併使用無水硫酸鈉乾燥,然後通過矽膠柱提純,所使用的淋洗液為甲醇:二氯甲烷:二乙胺=4:100:1(體積比),得到目標產物化合物a1,產率:24.8%。
化合物a1的確認:
1hnmr(200mhz,cdcl3)δ2.37(s,6h),3.59(s,2h),3.65-3.75(m,1h),3.80-3.92(m,3h),4.05-4.20(m,2h),4.46-4.50(m,1h),4.70-4.74(m,1h),6.68-6.72(m,3h),6.89-7.02(m,2h),7.10-7.17(m,1h),7.35-7.45(m,1h)。
lrms(m++1):理論值365.16,實測值:365.1674
實施例2
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-(2-氟乙氧基)-乙氧基)-乙氧基)-苯)硫醚(化合物a2)的合成
合成反應方程式:
將0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羥基)-苯)硫醚溶解在4mldmf中(hplc純),加入2倍摩爾量的碳酸鉀以及1-對甲苯磺醯基-(2-(2-氟乙氧基)-乙氧基)乙烷(1.2倍摩爾量),之後通入氮氣3-5min,將反應液在80-90℃條件下油浴加熱過夜。反應完後加入30ml水終止反應,並用乙酸乙酯萃取產物,合併有機相混合併使用無水硫酸鈉乾燥,然後通過矽膠柱提純,所使用的淋洗液為甲醇:二氯甲烷:二乙胺=4:100:1(體積比),得到目標產物化合物a2,產率:22.6%。
化合物a2的確認:
1hnmr(200mhz,cdcl3)δ2.37(s,6h),3.69-3.82(m,7h),3.83-3.88(m,3h),4.09-4.21(m,2h),4.44-4.48(m,1h),4.68-4.72(m,1h),6.68-6.74(m,3h),6.94-6.98(m,2h),7.12-7.22(m,1h),7.38-7.42(m,1h)。
lrms(m++1):理論值409.19,實測值409.1959。
實施例3
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(順-4-氟代-2-烯丁氧基)-苯)硫醚(化合物a3)的合成
合成反應方程式:
合成方法:
①1,4-二對甲苯磺醯基-2-丁烯的製備
將0.3ml的順-2-丁烯-1,4-二醇(3.16mmol)溶解在15ml新蒸四氫呋喃(thf)中,冰浴10min後,加入對甲苯磺醯氯1.76克(9.26mmol),冰浴下攪拌10min後,加入1克叔丁醇鈉,得到白色乳狀物,繼續攪拌過夜。反應完後用旋轉蒸發儀除去溶劑,加入20ml水,並用二氯甲烷萃取三次,合併有機相,經無水硫酸鈉乾燥後通過柱層析法提純,淋洗液為乙酸乙酯:石油醚=1:4(體積比),得到1,4-二對甲苯磺醯基-2-丁烯。產率:12.2%。
②1-氟代-4-對甲苯磺醯基-2-丁烯的製備
將1mmol1,4-二對甲苯磺醯基-2-丁烯加入兩口圓底燒瓶中,並加入3ml乙腈將其溶解,然後加入1ml1mol/l四正丁基氟化銨的四氫呋喃溶液(1mol/l溶液),通氮氣3min後將反應液在80℃條件下加熱30min,移除溶劑並且將所得紫色油狀混合物溶於10ml乙醚中,並加入10ml5%na2co3溶液,使粗產物在兩相間平衡10min。之後,使用乙醚進行萃取(10ml×2),合併有機相併用無水硫酸鈉乾燥,然後使用矽膠柱進行提純,淋洗液為乙酸乙酯:石油醚=1:4(體積比),得到1-氟代-4-對甲苯磺醯基-2-丁烯。產率:19.8%。
③2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(順-4-氟代-2-烯丁氧基)-苯)硫醚的製備
將0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羥基)-苯)硫醚溶解在4mldmf中(hplc純),加入2倍摩爾量的碳酸鉀以及1-氟代-4-對甲苯磺醯基-2-丁烯(1.2倍摩爾量),之後通入氮氣3-5min,將反應液在80-90℃條件下油浴加熱過夜。反應完後加入30ml水終止反應,並用乙酸乙酯萃取產物,合併有機相併使用無水硫酸鈉乾燥,然後通過矽膠柱提純,所使用的淋洗液為甲醇:二氯甲烷:二乙胺=4:100:1(體積比),得到目標產物化合物a3,產率:17%。
結構確認:
①化合物1,4-二對甲苯磺醯基-2-丁烯
1hnmr(400mhz,cdcl3):δ2.39(s,6h),4.47(d,j=5.85hz,4h),5.60-5.62(m,2h),7.28(d,j=8.08hz,4h),7.69(d,j=8.20hz,4h);
②化合物1-氟-4-對甲苯磺醯基-2-丁烯
1hnmr(400mhz,cdcl3):δ2.38(s,3h),4.57(d,j=6.6hz2h),4.83(dd,j=46.6,5.64hz,2h),5.58-5.64(m,1h),5.70-5.76(m,1h),7.28(d,j=8.12hz,2h),7.72(d,j=8.12hz,2h);
③化合物a3的確認:1hnmr(200mhz,cdcl3)δ2.34(s,6h),3.60(s,2h),4.75(dt,jfh=5.2hz,jhh=1.4hz,2h),4.99(dd,jfh=47hz,jhh=1.6hz,2h),5.88-5.91(m,2h)6.72-6.77(m,3h),6.90-6.98(m,2h),7.16-7.19(m,1h),7.42-7.46(m,1h).13cnmr(50mhz,cdcl3)δ43,62,65,81,114,116,116.5,117,126.5,127,129,129.5,130,132,135,148,157.
lrms(m++1):理論值:347.16,實測值:347.1592.
實施例4
二芳基硫醚衍生物的合成
2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(4-氟代-2-炔丁氧基)-苯)硫醚(化合物a4)的合成
合成反應方程式:
合成方法:
①化合物1,4-二對甲苯磺醯基-2-丁炔的製備
在圓底燒瓶中,將相應的10mmol的1,4-丁炔二醇(860mg)溶解在10ml的二氯甲烷中,冰浴降溫至0℃,逐滴加入1.4ml三乙胺(20mmol),以及20mg4-n,n-二甲基吡啶,之後繼續攪拌溶液5min;並將3.8gtscl溶解在3ml二氯甲烷中,通過恆壓滴液漏鬥逐滴加入到圓底燒瓶中,滴完後,反應液繼續在冰浴條件下攪拌1h,後於室溫反應3-4h;反應完後加入20ml二氯甲烷稀釋,並用1mol/lhcl溶液、5%na2co3溶液、飽和食鹽水依次洗滌。洗滌後的有機相經過無水硫酸鈉乾燥後,使用旋轉蒸發儀除去溶劑,並用矽膠柱提純,淋洗液為乙酸乙酯:石油醚=1:4(體積比),得到1,4-二對甲苯磺醯基-2-丁炔。
②化合物1-氟代-4-對甲苯磺醯基-2-丁炔的製備
將1mmol的1,4-二對甲苯磺醯基-2-丁炔加入到兩口圓底燒瓶中,並加入3ml乙腈將其溶解,然後加入1ml1mol/l四正丁基氟化銨的四氫呋喃溶液,通氮氣3min後,將反應液在80℃條件下加熱30min,移除溶劑並且將所得紫色油狀混合物溶於10ml乙醚,並加入10ml5%na2co3溶液,讓粗產物在兩相間平衡10min。之後,使用乙醚進行萃取(10ml×2),合併有機相併用無水硫酸鈉乾燥,使用矽膠柱提純產物,淋洗液為乙酸乙酯:石油醚=1:6(體積比),得到1-氟代-4-對甲苯磺醯基-2-丁炔。產率:42%。
③2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(4-氟代-2-炔丁氧基)-苯)硫醚的製備
將0.15-0.2mmol2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羥基)-苯)硫醚溶解在4mldmf中(hplc純),加入2倍摩爾量的碳酸鉀以及1-氟代-4-對甲苯磺醯基-2-丁炔(1.2倍摩爾量),之後通入氮氣3-5min,將反應液在80-90℃條件下油浴加熱過夜,反應完後加入30ml水終止反應,並用乙酸乙酯萃取產品,將有機相混合後使用無水硫酸鈉乾燥,並通過矽膠柱提純。淋洗液為甲醇:二氯甲烷:二乙胺=4:100:1(體積比),得到目標產物,產率:22.1%。
結構確認:
①化合物1,4-二對甲苯磺醯基-2-丁炔
1hnmr(400mhz,cdcl3):δ2.40(s,6h),4.51(s,4h),7.28(d,j=8.12hz,4h),7.69(d,j=8.24hz,4h);
②化合物1-氟代-4-對甲苯磺醯基-2-丁炔
1hnmr(400mhz,cdcl3):δ2.38(s,3h),4.67-4.70(m,2h),4.77(d,j=45.6hz,2h),7.29(d,j=8.04hz,2h),7.74(d,j=8.04hz,2h);
③化合物2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(4-氟代-2-炔丁氧基)-苯)硫醚
1hnmr(200mhz,cdcl3)δ2.34(s,6h),3.60(s,2h),4.72-4.77(m,2h),4.99(dd,jfh=47hz,jhh=1.6hz,2h),6.71-6.77(m,3h),6.91-6.98(m,2h),7.15-7.20(m,1h),7.42-7.46(m,1h);13cnmr(50mhz,cdcl3)δ43,56,62,68,72,85,115,115.5,117,119,128,130,136,138,148,155。
lrms(m++1):理論值345.13,實測值345.1391。
實施例5
[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-(2-氟乙氧基)-乙氧基)-乙氧基)-苯)硫醚(化合物a5)的合成
(1)[18f]-1-對甲苯磺醯基-2-((2-氟乙氧基)-乙氧基))乙烷的製備
乾燥[18f]kf/k222的準備:將水溶液中的[18f-]一次性通過qma小柱(4-(4-甲基哌啶)吡啶陰離子交換樹脂小柱)並推幹,後使用11mgk222/2mgk2co3溶液(0.8ml乙腈,0.2ml水)淋洗下放射性活度。之後將所得溶液在100℃條件下吹乾,並依次加入2ml無水乙腈(每次1ml)吹乾,得乾燥[18f]kf/k222。
將2mg的1-對甲苯磺醯基-2-((2-對甲苯磺醯基乙氧基)-乙氧基)-乙烷前體溶於1ml無水乙腈中,加入製備好的乾燥的[18f]kf/k222中。充分搖蕩均勻後置於85℃環境中加熱5min,停止加熱進行提純。
提純方式:反應液無需冷卻即加入5ml水,通過oasishlb(反相親水親脂小柱)柱子提純:首先將粗產品一次性以約每秒一滴的速度通過已經處理好的oasishlb柱子,使用5ml水洗柱子,吹乾,並且用氮氣流吹2min。之後用1.5ml乙腈直接淋洗產品至第二步反應管中(含有7.5mg氫化鈉,2mg第二步前體:2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羥基)-苯)硫醚的10ml試管)。
(2)[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(2-(2-(2-氟乙氧基)-乙氧基)-乙氧基)-苯)硫醚(化合物a5)的製備
在氮氣流條件下把上述加入了[18f]-1-對甲苯磺醯基-2-((2-氟乙氧基)-乙氧基))乙烷的試管中的溶液在120℃條件下吹乾。然後加入1ml無水dmf,搖勻後繼續在120℃條件下反應15min,反應結束使用oasishlb柱子初步提純產品:即先往反應粗產物中加入5ml水,將混合液一次性通過預先活化好的oasishlb柱子。使用5ml水淋洗柱子,推幹,並使用1ml乙醇淋洗產物,用hplc檢測(參見圖1),最終用半製備hplc分離純化即可得到18f標記目標產物[18f]-a5。
標記產率:標記總體時間約90min,標記中間體產率29.7%,最終產物產率:3.7%(按照投入的[18f-]的活度計算,產率未經時間校正),oasishlb柱子提純後產品放射化學純度為64%,主要放射性雜質是放射性中間體。
[18f]-a5的結構確證為將如上所述製得的標記產物[18f]-a5與實施例2製備的化合物a2即[19f]-a2,在相同的條件下共注射進行hplc分析,通過比對保留時間進行確認(參見圖2)。
分析用hplc條件如下:溶劑a:acn(乙腈),溶劑b:20mmafb(甲酸銨);梯度:0-3min95%b,3-11min95%-5%b,11-19min5%-95%b,19-21min95%b;柱子:gemini3micron,140x4.6mm,column:gemini3uc18,110a,150×4.6mm,3micron。
實施例6
[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(順-4-氟代-2-烯丁氧基)-苯)硫醚(化合物a6)的合成
(1)[18f]-1-氟代-4-對甲苯磺醯基-2-丁烯的製備
乾燥[18f]kf/k222的準備同實施例5。
將3mg的1,4-二對甲苯磺醯基-2-丁烯前體溶於1ml無水乙腈中,加入製備好的乾燥的[18f]kf/k222中。充分搖蕩均勻後置於60℃環境中加熱10min,停止加熱進行提純。
提純方式:使用0.5ml乙醚稀釋反應液。將反應液合併,並一次性通過矽膠小柱(sep-paksilica)後使用1.5ml乙醚將放射性中間體淋下。
(2)[18f]-2-氨基苯-(2-(n,n-二甲基)氨甲基-4-(順-4-氟代-2-烯丁氧基)-苯)硫醚(化合物a6)製備
在氮氣流條件下把加入了[18f]-1-氟代-4-對甲苯磺醯基-2-丁烯的試管中的乙醚在60℃條件下吹乾。然後加入1ml無水dmf,搖勻後,轉移到含有7.5mg碳酸銫,2mg2-氨基苯-(2-(n,n-二甲基)氨甲基-4-羥基)-苯)硫醚的試管中。混合均勻,並在20℃室溫下反應10min。反應結束使用oasishlb柱子初步提純產品:即先往反應粗產物中加入5ml水,將混合液一次性通過預先活化好的oasishlb柱子。使用5ml水淋洗柱子,推幹,並使用1ml50%乙醇/水溶液淋洗產物,用hplc檢測(圖3),[18f]-a6。
標記產率:標記總體時間約2h,以投入的[18f-]活度計算的不經時間校正產率:標記中間體產率約25%,[18f]-a6標記產率約1-2%。經過oasis小柱初步提純後的產品放射化學純度大於90%。
[18f]-a6的結構確證為將如上所述製得的標記產物[18f]-a6與實施例3製備的化合物a3即[19f]-a3,在相同的條件下共注射進行hplc分析,通過比對保留時間進行確認(參見圖3)。
分析用hplc條件如下:溶劑a:乙腈(acn),溶劑b:20mmafb(甲酸銨);梯度:0-3min95%b,3-11min95%-5%b,11-19min5%-95%b,19-21min95%b;柱子:gemini3micron,140x4.6mm,column:gemini3uc18,110a,150×4.6mm,3micron。
實施例7
體外競爭結合試驗(方法參見文獻shunichioya,j.med.chem.2002,45,4716-4723)。
體外競爭結合試驗中使用的三個測定非特異性結合的化合物如下:citalopram(西酞普蘭)、gbr12909(伐諾司林)和nisoxetine(尼索西汀),其中,citalopram是已知的sert抑制劑,gbr12909是已知的dat抑制劑,nisoxetine是已知的net抑制劑。
通過體外競爭實驗測得ki常數(數據見下表1),當二芳基硫醚化合物b環4位使用不同小分子鏈結合後,化合物保有對sert的高親和能力。由表1中的數據可知,本發明的化合物a1-a4均對sert有非常高的親和性,尤其是化合物a1(0.09±0.01nm)、a3(0.02±0.003nm)。而且,對dat和net的結合試驗顯示,這些化合物對dat的親和性很差,對net的親和性也一般,從而表現出了高選擇性。例如,化合物a3的dat/sert選擇性高達34300,net/sert選擇性也高達4010,利用18f標記後可有望作為sert顯像劑。
表1:二芳基硫醚系列衍生化合物與sert、dat、net受體的競爭性結合常數kia
a.每個ki值都是三次實驗所得。
b.sert結合性能測定實驗中,llc-sert細胞膜勻漿與0.1nm[125i]idam(kd=0.2nm);dat結合性能測定實驗中,llc-dat細胞膜勻漿與0.08nm[125i]ipt(kd=1.2);net結合性能測定實驗中,llc-net細胞膜勻漿與0.06nm[125i]2-inxt(kd=0.06nm)。
c.n=1。
上述用於放射性受體結合實驗的標準化合物化學結構如下: