一種能改善捲菸抽吸效果的異黃酮類化合物及其製備方法與應用與流程
2023-05-29 10:24:31 3
本發明屬於植物化學
技術領域:
,具體涉及一種從中藥材葛根中首次提取得到的異黃酮類化合物及其製備方法和應用。該化合物可作為捲菸嘴棒添加劑用於改善捲菸抽吸效果中。
背景技術:
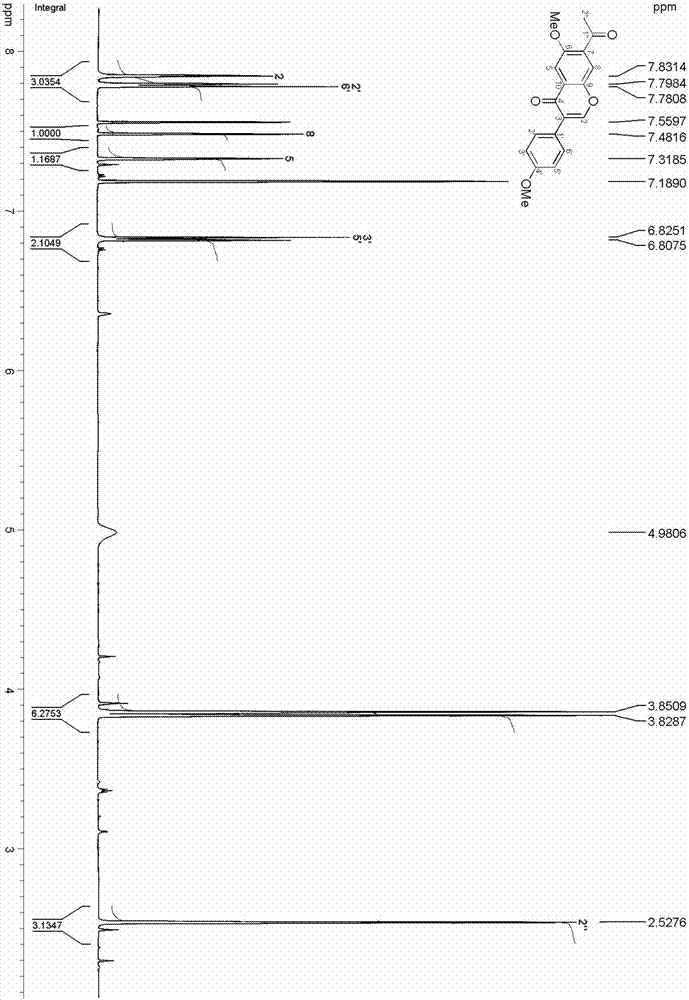
:葛根為豆科植物野葛的乾燥根,習稱野葛。秋、冬二季採挖,趁鮮切成厚片或小塊;乾燥。甘、辛,涼。有解肌退熱,透疹,生津止渴,昇陽止瀉之功。常用於表證發熱,項背強痛,麻疹不透,熱病口渴,陰虛消渴,熱瀉熱痢,脾虛洩瀉。同時葛根也是一種重要的保健食品,其藥用價值極高,素有「亞洲人參」之美譽,葛粉稱之為「長壽粉」,在日本被譽為「皇室特供食品」。葛根中的主要化學成分為黃酮和異黃酮類化合物(大豆素、大豆甙、葛根素、葛根素-7-木糖甙等),也含有萜類、內酯、甾醇等其它結構類型的化合物。異黃酮是黃酮類化合物中的一種,它是植物苯丙氨酸代謝過程中,由肉桂醯輔酶a側鏈延長後環化形成以苯色酮環為基礎的酚類化合物,其3-苯基衍生物即為異黃酮類化合物。異黃酮類化合物突出的生理活性引起了科技工作者的廣泛關注。研究表明葛根異黃酮具有補充女性雌激素、收縮平滑肌、增加冠血流量、抑制血小板凝集、降血糖等作用;異黃酮還具有抗氧化、抗腫瘤、預防動脈硬化、改善骨質疏鬆等其他多種功效。此外,異黃酮類化合物還有改善人味覺效果的功能,在食品工業中也有廣泛的應用。本發明從葛根中分離得到了一種異黃酮類化合物,該化合物添加到捲菸過濾嘴中,能顯著降低捲菸抽吸喉部刺激性,具有明顯的潤喉功效。本發明化合物添加到捲菸濾嘴中,能明顯改善捲菸抽吸舒適性。和對照相比較,捲菸抽吸的潤感有提升、生津作用明顯,抽吸的喉部舒適性得到明顯改善,目前尚未發現相關報導。技術實現要素:本發明的第一目的在於提供一種異黃酮類化合物;第二目的在於提供所述異黃酮類化合物的製備方法;第三目的在於提供所述異黃酮類化合物作為改善捲菸抽吸舒適性添加劑的應用,添加本發明化合物後,捲菸抽吸的潤感有提升、生津作用明顯,抽吸喉部舒適性得到明顯改善。為實現上述目的,本發明採用的技術方案如下:本發明的第一目的是這樣實現的,所述的異黃酮類化合物是從傳統中藥材葛根中分離得到,其分子式為c19h16o5,其結構式如式(i)所示:該化合物為淺黃色膠狀物,命名為:7-乙醯基-4′,6-二甲氧基-異黃酮,英文名為:7-acetyl-4′,6-dimethoxy-isoflavone。本發明的第二目的是這樣實現的,所述異黃酮類化合物的製備方法,以葛根為原料,經浸膏提取、有機溶劑萃取、mci脫色、矽膠柱層析和高效液相色譜分離步驟製得,具體為:a、浸膏提取:將葛根粉碎到20~40目,用溶劑超聲提取2~5次,每次所用提取溶劑的質量為葛根質量的2~4倍,每次30~60分鐘,合併提取液並過濾,濾液減壓濃縮至肉眼觀察到剛有沉澱析出,靜置3~5h,濾除沉澱物,之後將所得濾液濃縮成浸膏a;b、有機溶劑萃取:向浸膏a中加入重量是浸膏a重量1~2倍的水,然後用有機溶劑萃取3~5次,每次所用有機溶劑的體積與水體積的相同,合併有機溶劑萃取相,之後將合併得到的有機溶劑萃取相減壓濃縮成浸膏b;c、mci脫色:向浸膏b中加入是浸膏b重量3~5倍的純甲醇,待浸膏b完全溶解後,上mci柱,用體積濃度為90~95%甲醇水溶液進行洗脫,合併洗脫液,之後將合併後的洗脫液減壓濃縮成浸膏c;d、矽膠柱層析:將浸膏c上矽膠柱層析,裝柱矽膠為160~200目,所用矽膠的重量為浸膏c重量6~10倍量;以體積比為1:0~0:1的氯仿和丙酮混合有機溶劑梯度洗脫,收集各梯度的梯度洗脫液並濃縮,經tlc監測,合併相同的部分;每個梯度洗脫到tlc點板無點後(即該梯度洗脫不出物質後),更換下一梯度洗脫;e、高效液相色譜分離:將採用體積比為7:3的氯仿-丙酮混合有機溶劑洗脫得到部分採用高效液相色譜分離純化,即得所述的異黃酮類化合物。進一步,優選的是,所述a步驟的溶劑為體積濃度為70~100%的丙酮水溶液、體積濃度為90~100%的乙醇水溶液或體積濃度為90~100%的甲醇水溶液。進一步,優選的是,所述b步驟的有機溶劑為二氯甲烷、氯仿、乙酸乙酯、乙醚或石油醚。進一步,優選的是,所述d步驟中浸膏c在經矽膠柱層析前,先用重量是浸膏c1.5~3倍的丙酮或者甲醇溶解,然後用重量是浸膏c0.8~1.2倍的80~100目矽膠拌樣,之後上樣。進一步,優選的是,所述d步驟中,梯度洗脫時,所使用的氯仿和丙酮混合有機溶劑的體積比依次為20:1、9:1、8:2、7:3、6:4和1:1;每個梯度洗脫到tlc點板無點後,更換下一梯度洗脫。進一步,優選的是,所述e步驟的高效液相色譜分離純化是以體積濃度為50%的甲醇水溶液為流動相,流速15~25ml/min,以21.2×250mm,5μm的zorbaxprephtgf反相製備柱為固定相,紫外檢測器檢測波長為358nm,每次進樣10~100μl,收集39.2min的色譜峰,多次累加後蒸乾。本領域技術人員應該知曉本技術方案只是一個優選技術方案,高效液相色譜分離純化採用的流動相不限於此。以上述方法製備得到的異黃酮類化合物的結構通過以下方法進行測定:本發明化合物為黃色膠狀物;hresi-ms顯示其準分子離子峰為347.0890[m+na]+(計算值347.0895),結合1hnmr和dept譜確定其分子式為c19h16o5,不飽和度為12。紅外光譜中顯示了羰基(1715和1656cm-1)和芳環(1610、1519和1436cm-1)的共振吸收峰。而紫外光譜在210、256、312和358nm有最大吸收也說明了化合物中可能存在芳環結構。化合物的1h和13cnmr譜(如表1、圖1和圖2)顯示其含有19個碳和16個氫,包括1個異黃酮骨架(c-1~c-10和c-1′~c-6′,h-5、h-8、h-2′,6′和h-2′,6′),二個甲氧基(δc55.9q和56.3q,δh3.82s和3.85s)和一個乙醯基(δc198.6s和29.7q,δh2.53s)。化合物的異黃酮骨架可進一步由h-5和c-4、c-6、c-7、c-9、c-10,h-8和c-1″、c-6、c-7、c-9、c-10,h-2和c-1′、c-3、c-4、c-9,以及h-2′,6′和c-3的hmbc相關得到確認。進一步分析其hmbc相關譜(如圖3),根據兩個甲氧基氫(δh3.82,3.85)與c-6(δc155.0)和c-4′(δc160.3)的hmbc相關可推測兩個甲氧基取代在異黃酮母核的c-6和c-4′位。乙醯基取代在c-7位可由h-2″(δh2.53)與c-7(δc125.9),以及h-8(δh7.48)與c-1″(δc198.6)的hmbc相關得到確定。苯環上典型的質子信號h-5(δh7.32)、h-8(δh7.48)、h-2,6[δh7.88(d,j=8.8)]和h-3,5[(δh6.81(d,j=8.8)]也支持異黃酮母核上的上述取代基模式。至此,化合物的結構得到確定,並命名為化合物命名為:7-乙醯基-4′,6-二甲氧基-異黃酮。化合物的紅外、紫外和質譜數據:uv(甲醇),λmax(logε)358(3.62)、312(3.09)、256(3.97)、210(4.32)nm;ir(溴化鉀壓片):νmax3087、2926、1715、1656、1610、1562、1519、1436、1368、1269、1142、1065、908、820cm-1;1h和13cnmr數據(500和125mhz,(c5d5n),見表1;正離子模式esimsm/z347[m+na]+;正離子模式hresimsm/z347.0890[m+na]+(計算值c19h16o5,347.0895)。表1.本發明化合物的1hnmr和13cnmr數據(c5d5n)no.δcδhno.δcδh2152.7d7.83s1′124.2s3122.5s2′6′129.3d7.88(d)8.84175.8s3′5′115.1d6.81(d)8.85115.1d7.32s4′160.3s6155.0s1″198.6s7125.9s2″29.7q2.53s8118.1d7.48some-4′55.9q3.82s9151.0some-656.3q3.85s10130.2s本發明的第三目的是這樣實現的:所述的異黃酮類化合物作為製備捲菸濾嘴添加劑的應用。考慮到三醋酸甘油酯為捲菸濾嘴成型最常用的增塑劑,而且本發明化合物溶於三醋酸甘油酯,在捲菸濾嘴成型過程中,本發明化合物通過三醋酸甘油酯添加到過濾嘴中。用三醋酸甘油酯將上述異黃酮化合物用配成0.1mg/ml的溶液。按濾嘴絲束重量的8%均勻的噴灑到濾嘴絲束上,製成濾棒,然後將該濾棒通過常規的捲菸卷接製成捲菸。本發明與現有技術相比,其有益效果為:本發明異黃酮類化合物是首次被分離出來的,通過核磁共振和質譜測定方法確定為異黃酮類化合物,並表徵了其結構。本發明化合物通過三醋酸甘油酯添加到捲菸過濾嘴中,以未添加該化合物的相同捲菸作為對照,進行感官評析。評析結果表明:和對照相比較,添加本發明化合物能降低捲菸抽吸喉部刺激性,具有明顯改善捲菸抽吸品質效果。本發明化合物添加到捲菸濾嘴中,工藝上容易實現,不增加生產過程中的額外步驟,可明顯提升捲菸抽吸品質。而且本發明化合物結構簡單,從天然植物中容易提取,通過人工合成在工藝上也容易實現,生產成本低,捲菸添加效果好,具有良好的應用前景。附圖說明圖1為本發明異黃酮類化合物的核磁共振碳譜(13cnmr);圖2為本發明異黃酮類化合物的核磁共振氫譜(1hnmr);圖3本發明異黃酮類化合物的關鍵hmbc相關圖。具體實施方式下面結合實施例對本發明作進一步的詳細描述。本領域技術人員將會理解,下列實施例僅用於說明本發明,而不應視為限定本發明的範圍。實施例中未註明具體技術或條件者,按照本領域內的文獻所描述的技術或條件或者按照產品說明書進行。所用試劑或儀器未註明生產廠商者,均為可以通過購買獲得的常規產品。本發明所採用的葛根全部是普通市購幹製品。本發明所述的異黃酮類化合物,是從傳統中藥材葛根中分離得到,其分子式為c19h16o5,其結構式如式(i)所示:命名為:化合物命名為:7-乙醯基-4′,6-二甲氧基-異黃酮,英文名為:7-acetyl-4′,6-dimethoxy-isoflavone。本發明所述異黃酮類化合物的製備方法,以葛根為原料,經浸膏提取、有機溶劑萃取、mci脫色、矽膠柱層析和高效液相色譜分離步驟製得,具體為:a、浸膏提取:將葛根粉碎到20~40目,用溶劑超聲提取2~5次,每次所用提取溶劑的質量為葛根質量的2~4倍,每次30~60分鐘,合併提取液並過濾,濾液減壓濃縮至肉眼觀察到剛有沉澱析出,靜置3~5h,濾除沉澱物,之後將所得濾液濃縮成浸膏a;b、有機溶劑萃取:向浸膏a中加入重量是浸膏a重量1~2倍的水,然後用有機溶劑萃取3~5次,每次所用有機溶劑的體積與水體積的相同,合併有機溶劑萃取相,之後將合併得到的有機溶劑萃取相減壓濃縮成浸膏b;c、mci脫色:向浸膏b中加入是浸膏b重量3~5倍的純甲醇,待浸膏b完全溶解後,上mci柱,用體積濃度為90~95%甲醇水溶液進行洗脫,合併洗脫液,之後將合併後的洗脫液減壓濃縮成浸膏c;d、矽膠柱層析:將浸膏c上矽膠柱層析,裝柱矽膠為160~200目,所用矽膠的重量為浸膏c重量6~10倍量;以體積比為1:0~0:1的氯仿和丙酮混合有機溶劑梯度洗脫,收集各梯度的梯度洗脫液並濃縮,經tlc監測,合併相同的部分;每個梯度洗脫到tlc點板無點後(即該梯度洗脫不出物質後),更換下一梯度洗脫;e、高效液相色譜分離:將採用體積比為7:3的氯仿-丙酮混合有機溶劑洗脫得到部分採用高效液相色譜分離純化,即得所述的異黃酮類化合物。所述a步驟的溶劑為體積濃度為70~100%的丙酮水溶液、體積濃度為90~100%的乙醇水溶液或體積濃度為90~100%的甲醇水溶液。所述b步驟的有機溶劑為二氯甲烷、氯仿、乙酸乙酯、乙醚或石油醚。所述d步驟中浸膏c在經矽膠柱層析前,先用重量是浸膏c1.5~3倍的丙酮或者甲醇溶解,然後用重量是浸膏c0.8~1.2倍的80~100目矽膠拌樣,之後上樣。所述d步驟中,梯度洗脫時,所使用的氯仿和丙酮混合有機溶劑的體積比依次為20:1、9:1、8:2、7:3、6:4和1:1;每個梯度洗脫到tlc點板無點後,更換下一梯度洗脫。所述e步驟的高效液相色譜分離純化是以體積濃度為50%的甲醇水溶液為流動相,流速15~25ml/min,以21.2×250mm,5μm的zorbaxprephtgf反相製備柱為固定相,紫外檢測器檢測波長為358nm,每次進樣10~100μl,收集39.2min的色譜峰,多次累加後蒸乾。實施例1a、浸膏提取:將葛根粉碎到20目,用溶劑超聲提取2次,每次所用提取溶劑的質量為葛根質量的2倍,每次30分鐘,合併提取液並過濾,濾液減壓濃縮至肉眼觀察到剛有沉澱析出,靜置3h,濾除沉澱物,之後將所得濾液濃縮成浸膏a;b、有機溶劑萃取:向浸膏a中加入重量是浸膏a重量1倍的水,然後用有機溶劑萃取3次,每次所用有機溶劑的體積與水體積的相同,合併有機溶劑萃取相,之後將合併得到的有機溶劑萃取相減壓濃縮成浸膏b;c、mci脫色:向浸膏b中加入是浸膏b重量3倍的純甲醇,待浸膏b完全溶解後,上mci柱,用體積濃度為90%甲醇水溶液進行洗脫,合併洗脫液,之後將合併後的洗脫液減壓濃縮成浸膏c;d、矽膠柱層析:浸膏c先用重量是浸膏c1.5倍的丙酮溶解,然後用重量是浸膏c0.8倍的80目矽膠拌樣,之後上樣進行柱層析,其中,裝柱矽膠為160目,所用矽膠的重量為浸膏c重量6倍量;以體積比依次為20:1、9:1、8:2、7:3、6:4和1:1的氯仿和丙酮混合有機溶劑梯度洗脫,收集各梯度的梯度洗脫液並濃縮,經tlc監測,合併相同的部分;每個梯度洗脫到tlc點板無點後(即該梯度洗脫不出物質後),更換下一梯度洗脫;e、高效液相色譜分離:將採用體積比為7:3的氯仿-丙酮混合有機溶劑洗脫得到部分採用高效液相色譜分離純化,高效液相色譜分離純化的具體方法是:以體積濃度為50%的甲醇水溶液為流動相,流速15ml/min,以21.2×250mm,5μm的zorbaxprephtgf反相製備柱為固定相,紫外檢測器檢測波長為358nm,每次進樣10μl,收集39.2min的色譜峰,多次累加後蒸乾,即得所述的異黃酮類化合物。其中,所述a步驟的溶劑為體積濃度為80%的丙酮水溶液。所述b步驟的有機溶劑為二氯甲烷。實施例2a、浸膏提取:將葛根粉碎到40目,用溶劑超聲提取5次,每次所用提取溶劑的質量為葛根質量的4倍,每次60分鐘,合併提取液並過濾,濾液減壓濃縮至肉眼觀察到剛有沉澱析出,靜置5h,濾除沉澱物,之後將所得濾液濃縮成浸膏a;b、有機溶劑萃取:向浸膏a中加入重量是浸膏a重量2倍的水,然後用有機溶劑萃取5次,每次所用有機溶劑的體積與水體積的相同,合併有機溶劑萃取相,之後將合併得到的有機溶劑萃取相減壓濃縮成浸膏b;c、mci脫色:向浸膏b中加入是浸膏b重量5倍的純甲醇,待浸膏b完全溶解後,上mci柱,用體積濃度為95%甲醇水溶液進行洗脫,合併洗脫液,之後將合併後的洗脫液減壓濃縮成浸膏c;d、矽膠柱層析:浸膏c先用重量是浸膏c3倍的甲醇溶解,然後用重量是浸膏c.2倍的100目矽膠拌樣,之後上樣進行柱層析,其中,裝柱矽膠為200目,所用矽膠的重量為浸膏c重量10倍量;以體積比依次為20:1、9:1、8:2、7:3、6:4和1:1的氯仿和丙酮混合有機溶劑梯度洗脫,收集各梯度的梯度洗脫液並濃縮,經tlc監測,合併相同的部分;每個梯度洗脫到tlc點板無點後(即該梯度洗脫不出物質後),更換下一梯度洗脫;e、高效液相色譜分離:將採用體積比為7:3的氯仿-丙酮混合有機溶劑洗脫得到部分採用高效液相色譜分離純化,高效液相色譜分離純化的具體方法是:以體積濃度為50%的甲醇水溶液為流動相,流速125ml/min,以21.2×250mm,5μm的zorbaxprephtgf反相製備柱為固定相,紫外檢測器檢測波長為358nm,每次進樣100μl,收集39.2min的色譜峰,多次累加後蒸乾,即得所述的異黃酮類化合物。其中,所述a步驟的溶劑為丙酮。所述b步驟的有機溶劑為乙醚。實施例3a、浸膏提取:將葛根粉碎到30目,用溶劑超聲提取3次,每次所用提取溶劑的質量為葛根質量的3倍,每次40分鐘,合併提取液並過濾,濾液減壓濃縮至肉眼觀察到剛有沉澱析出,靜置4h,濾除沉澱物,之後將所得濾液濃縮成浸膏a;b、有機溶劑萃取:向浸膏a中加入重量是浸膏a重量1.5倍的水,然後用有機溶劑萃取4次,每次所用有機溶劑的體積與水體積的相同,合併有機溶劑萃取相,之後將合併得到的有機溶劑萃取相減壓濃縮成浸膏b;c、mci脫色:向浸膏b中加入是浸膏b重量4倍的純甲醇,待浸膏b完全溶解後,上mci柱,用體積濃度為92%甲醇水溶液進行洗脫,合併洗脫液,之後將合併後的洗脫液減壓濃縮成浸膏c;d、矽膠柱層析:浸膏c先用重量是浸膏c2倍的丙酮溶解,然後用重量是浸膏c1倍的90目矽膠拌樣,之後上樣進行柱層析,其中,裝柱矽膠為180目,所用矽膠的重量為浸膏c重量8倍量;以體積比依次為20:1、9:1、8:2、7:3、6:4和1:1的氯仿和丙酮混合有機溶劑梯度洗脫,收集各梯度的梯度洗脫液並濃縮,經tlc監測,合併相同的部分;每個梯度洗脫到tlc點板無點後(即該梯度洗脫不出物質後),更換下一梯度洗脫;e、高效液相色譜分離:將採用體積比為7:3的氯仿-丙酮混合有機溶劑洗脫得到部分採用高效液相色譜分離純化,高效液相色譜分離純化的具體方法是:以體積濃度為50%的甲醇水溶液為流動相,流速22ml/min,以21.2×250mm,5μm的zorbaxprephtgf反相製備柱為固定相,紫外檢測器檢測波長為358nm,每次進樣60μl,收集39.2min的色譜峰,多次累加後蒸乾,即得所述的異黃酮類化合物。其中,體積濃度為90%的乙醇水溶液;所述b步驟的有機溶劑為石油醚。實施例4a、浸膏提取:將4.4kg葛根粉碎到30目,用溶劑超聲提取4次,每次所用提取溶劑的質量為葛根質量的3倍,每次60分鐘,合併提取液並過濾,濾液減壓濃縮至肉眼觀察到剛有沉澱析出,靜置4h,濾除沉澱物,之後將所得濾液濃縮成浸膏a,浸膏a重量為120g;b、有機溶劑萃取:向浸膏a中加入重量是浸膏a重量2倍的水,然後用有機溶劑萃取5次,每次所用有機溶劑的體積與水體積的相同,合併有機溶劑萃取相,之後將合併得到的有機溶劑萃取相減壓濃縮成浸膏b,浸膏b重量為80g;c、mci脫色:向浸膏b中加入是浸膏b重量3倍的純甲醇,待浸膏b完全溶解後,上mci柱,用體積濃度為90%甲醇水溶液進行洗脫,合併洗脫液,之後將合併後的洗脫液減壓濃縮成浸膏c,浸膏c重量為62g;d、矽膠柱層析:浸膏c先用重量120g的丙酮溶解,然後用重量為62g的100目矽膠拌樣,之後上樣進行柱層析,其中,裝柱矽膠為200目,所用矽膠的重量為400g;以體積比依次為20:1、9:1、8:2、7:3、6:4和1:1的氯仿和丙酮混合有機溶劑梯度洗脫,收集各梯度的梯度洗脫液並濃縮,經tlc監測,合併相同的部分,得到6個部分a-f;每個梯度洗脫到tlc點板無點後(即該梯度洗脫不出物質後),更換下一梯度洗脫;e、高效液相色譜分離:將採用體積比為7:3的氯仿-丙酮混合有機溶劑洗脫得到部分(d部分12g)採用高效液相色譜分離純化,高效液相色譜分離純化的具體方法是:以體積濃度為50%的甲醇水溶液為流動相,流速20ml/min,以21.2×250mm,5μm的zorbaxprephtgf反相製備柱為固定相,紫外檢測器檢測波長為358nm,每次進樣50μl,收集39.2min的色譜峰,多次累加後蒸乾,即得所述的異黃酮類化合物。其中,所述a步驟的溶劑為體積濃度為70%的丙酮水溶液。所述b步驟的有機溶劑為氯仿。實施例5a、浸膏提取:將10kg葛根粉碎到40目,用溶劑超聲提取4次,每次所用提取溶劑的質量為葛根質量的2.5倍,每次48分鐘,合併提取液並過濾,濾液減壓濃縮至肉眼觀察到剛有沉澱析出,靜置4.5h,濾除沉澱物,之後將所得濾液濃縮成浸膏a,浸膏a重量為300g;b、有機溶劑萃取:向浸膏a中加入重量為350g的水,然後用有機溶劑萃取5次,每次所用有機溶劑的體積與水體積的相同,合併有機溶劑萃取相,之後將合併得到的有機溶劑萃取相減壓濃縮成浸膏b,浸膏b重量為210g;c、mci脫色:向浸膏b中加入是浸膏b重量3.5倍的純甲醇,待浸膏b完全溶解後,上mci柱,用體積濃度為90%甲醇水溶液15l進行洗脫,合併洗脫液,之後將合併後的洗脫液減壓濃縮成浸膏c,浸膏c重量為150g;d、矽膠柱層析:浸膏c先用重量是浸膏c2倍的丙酮溶解,然後用重量是浸膏c1倍的100目矽膠拌樣,之後上樣進行柱層析,其中,裝柱矽膠為200目,所用矽膠的重量為浸1000g;以體積比依次為20:1、9:1、8:2、7:3、6:4和1:1的氯仿和丙酮混合有機溶劑梯度洗脫,收集各梯度的梯度洗脫液並濃縮,經tlc監測,合併相同的部分,得到6個部分a-f;每個梯度洗脫到tlc點板無點後(即該梯度洗脫不出物質後),更換下一梯度洗脫;e、高效液相色譜分離:將採用體積比為7:3的氯仿-丙酮混合有機溶劑洗脫得到部分(d部分32g)採用高效液相色譜分離純化,高效液相色譜分離純化的具體方法是:以體積濃度為50%的甲醇水溶液為流動相,流速20ml/min,以21.2×250mm,5μm的zorbaxprephtgf反相製備柱為固定相,紫外檢測器檢測波長為358nm,每次進樣80μl,收集39.2min的色譜峰,多次累加後蒸乾,即得所述的異黃酮類化合物。所述a步驟的溶劑為甲醇。所述b步驟的有機溶劑為乙酸乙酯。實施例6將實施例1製備得到的異黃酮類化合物的結構通過以下方法進行測定:本發明化合物為黃色膠狀物;hresi-ms顯示其準分子離子峰為347.0890[m+na]+(計算值347.0895),結合1hnmr和dept譜確定其分子式為c19h16o5,不飽和度為12。紅外光譜中顯示了羰基(1715和1656cm-1)和芳環(1610、1519和1436cm-1)的共振吸收峰。而紫外光譜在210、256、312和358nm有最大吸收也說明了化合物中可能存在芳環結構。化合物的1h和13cnmr譜(如表1、圖1和圖2)顯示其含有19個碳和16個氫,包括1個異黃酮骨架(c-1~c-10和c-1′~c-6′,h-5、h-8、h-2′,6′和h-2′,6′),二個甲氧基(δc55.9q和56.3q,δh3.82s和3.85s)和一個乙醯基(δc198.6s和29.7q,δh2.53s)。化合物的異黃酮骨架可進一步由h-5和c-4、c-6、c-7、c-9、c-10,h-8和c-1″、c-6、c-7、c-9、c-10,h-2和c-1′、c-3、c-4、c-9,以及h-2′,6′和c-3的hmbc相關得到確認。進一步分析其hmbc相關譜(如圖3),根據兩個甲氧基氫(δh3.82,3.85)與c-6(δc155.0)和c-4′(δc160.3)的hmbc相關可推測兩個甲氧基取代在異黃酮母核的c-6和c-4′位。乙醯基取代在c-7位可由h-2″(δh2.53)與c-7(δc125.9),以及h-8(δh7.48)與c-1″(δc198.6)的hmbc相關得到確定。苯環上典型的質子信號h-5(δh7.32)、h-8(δh7.48)、h-2,6[δh7.88(d,j=8.8)]和h-3,5[(δh6.81(d,j=8.8)]也支持異黃酮母核上的上述取代基模式。至此,化合物的結構得到確定,並命名為化合物命名為:7-乙醯基-4′,6-二甲氧基-異黃酮。實施例7取實施例2-5製備的化合物,為黃色膠狀物。測定方法與實施例6相同,確認實施例2-5製備的化合物為所述異黃酮類化合物——7-乙醯基-4′,6-二甲氧基-異黃酮。實施例8取實施例1-5中任一製備的異黃酮化合物進行捲菸濾嘴的添加效果試驗,試驗情況如下:供添加用捲菸為紅雲紅河集團的捲菸「紫雲」,用三醋酸甘油酯將上述異黃酮化合物用配成0.1mg/ml的溶液。按濾嘴絲束重量的8%均勻的噴灑到濾嘴絲束上,製成濾棒,然後將該濾棒通過常規的捲菸卷接製成捲菸,進行感官評析,並以未添加該化合物的相同捲菸作為對照。評析結果表明:和對照相比較,捲菸抽吸的潤感有提升、生津作用明顯,捲菸抽吸舒適性得到明顯改善,結果見表2。實施例9取實施例1-5中任一製備的異黃酮化合物進行捲菸濾嘴的添加效果試驗,供添加用捲菸為紅雲紅河集團的捲菸「紫雲」,用三醋酸甘油酯將上述異黃酮化合物用配成0.2mg/ml的溶液。按濾嘴絲束重量的5%均勻的噴灑到濾嘴絲束上,製成濾棒,然後將該濾棒通過常規的捲菸卷接製成捲菸,進行感官評析,並以未添加該化合物的相同捲菸作為對照。評析結果表明:和對照相比較,捲菸抽吸的潤感有提升、生津作用明顯,捲菸抽吸喉部舒適性得到明顯改善,結果見表2。表2捲菸感官舒適度評價註:分數越高,產品感官質量越好。本發明化合物應用方法不限於此,還可添加至內襯紙上等。以上顯示和描述了本發明的基本原理、主要特徵和本發明的優點。本行業的技術人員應該了解,本發明不受上述實施例的限制,上述實施例和說明書中描述的只是說明本發明的原理,在不脫離本發明精神和範圍的前提下,本發明還會有各種變化和改進,這些變化和改進都落入要求保護的本發明範圍內。本發明要求保護範圍由所附的權利要求書及其等效物界定。當前第1頁12