一種複合發泡劑及其製備方法與流程
2023-05-30 01:28:31 3
本發明涉及發泡技術領域,尤其涉及一種複合發泡劑及其製備方法。
背景技術:
微孔發泡塑料是一種泡孔尺寸在0.1~10μm之間,泡孔密度在109~1015cells/cm3範圍內的具有閉孔結構的新型多孔材料。微孔發泡最初是由美國麻省理工學院的Suh教授提出,其基本設計思想是如果泡孔尺寸比聚合物中的裂紋尺寸還小,則微孔不僅不會降低材料的性能,反而可以鈍化裂紋尖端,能夠有效阻止裂紋擴展;此外,微孔還能減緩聚合物材料內部的應力集中,改善和提高抗衝擊性能。事實證明,與未發泡材料相比,微孔發泡材料具有密度低、衝擊強度高、韌性好、比強度高、疲勞壽命長等優異性能,被稱為「21世紀的新型材料」,具有較好的應用前景。
對於微孔發泡塑料或微孔發泡技術而言,發泡劑起著至關重要的作用。製備發泡塑料的常用發泡劑有兩種類型:物理髮泡劑和化學發泡劑,採用物理髮泡劑需要結合超臨界流體技術,其繁瑣的工藝和高昂的成本使其在實際生產中難以得到廣泛應用,相比之下,化學發泡劑使用起來較為簡單方便,成為製備微孔發泡塑料的首選。
例如,偶氮二甲醯胺(AC)發泡劑便是塑料加工業中常用的一種化學發泡劑,分解時主要釋放出N2、CO和少量CO2,發氣量大,不助燃且有自熄性,無毒,無汙染且價格便宜,適用於聚乙烯、聚丙烯、聚氯乙烯、乙烯-乙酸乙烯共聚物、ABS等樹脂的常壓發泡、加壓發泡或擠出發泡等。然而,AC發泡劑顆粒較粗,容易團聚,難以在樹脂基體中均勻分散,給發泡工藝造成困難,且影響發泡製品的質量,容易使發泡製品表面泛黃或噴霜。
針對於此,現有技術主要通過細化AC顆粒來改善其使用效果,且通常通過以下四種方式來細化AC顆粒:(1)氣流粉碎法;(2)通過添加尿素、表面活性劑或者藉助是超聲處理等氧化工藝進行優化;(3)氯酸鈉氧化法;(4)重結晶法。但是,通過以上四種方法獲得的AC顆粒粒徑仍然較粗,為微米級別,其改善作用並不明顯。
為了進一步細化AC顆粒,現有技術還提出了一些複合型發泡劑,如公開號為CN102250374A的專利公開了一種基於金屬-有機配位聚合物的複合發泡劑,以金屬-有機物為多孔基體,將發泡劑組裝進入基體的孔道內來細化發泡劑顆粒,防止團聚;公開號為CN102250375A的專利公開了一種基於多孔無機材料的複合發泡劑,其採用多孔無機材料(分子篩、活性炭、無機氣凝膠、無機氧化物凝膠、矽膠)和非矽基多孔無機材料為載體,同樣是將發泡劑組裝到多孔材料的孔道中來細化發泡劑;公開號為CN102898672A公開了一種微孔發泡塑料發泡劑,以鎢酸鈉改性的凹凸棒土為載體,將AC發泡劑吸附負載於載體上,能夠分散細化AC顆粒。然而,上述複合發泡劑中發泡劑在載體上的負載量較低,使用時需投入較大的量,使用成本較高。另外,上述複合發泡劑中的載體為改性載體,製備過程中需要利用複雜的改性工藝來改性載體材料,在形成改性的載體後,才能作為可用載體進一步與發泡劑複合,製備工序複雜,成本較高。
技術實現要素:
有鑑於此,本發明的目的在於提供一種複合發泡劑及其製備方法,本發明的複合發泡劑具有高負載量,有利於降低使用成本。另外,本發明提供的製備方法簡便易行,便於生產應用。
本發明提供了一種複合發泡劑,包括磷酸鋯載體和負載於所述磷酸鋯載體上的偶氮二甲醯胺。
優選的,所述磷酸鋯為α-磷酸鋯。
優選的,所述磷酸鋯的片層厚度為5~20nm。
優選的,所述偶氮二甲醯胺分散於磷酸鋯的片層之間和/或吸附於磷酸鋯的片層表面。
優選的,所述偶氮二甲醯胺與磷酸鋯載體的質量比為(0.25~2)∶1。
本發明還提供了上述技術方案所述複合發泡劑的製備方法,包括以下步驟:
A)將偶氮二甲醯胺與溶劑混合,形成發泡劑溶液;
B)將磷酸鋯與所述發泡劑溶液混合,過濾,得到複合發泡劑。
優選的,所述溶劑為二甲基亞碸和/或二甲基甲醯胺。
優選的,所述步驟A)中,偶氮二甲醯胺的質量與溶劑的體積比為(1~10)g:(10~500)mL。
優選的,所述步驟A)中,所述混合的溫度為30~80℃;所述步驟B)中,所述混合的溫度為40~100℃。
優選的,所述偶氮二甲醯胺與磷酸鋯的質量比為(0.25~2)∶1。
與現有技術相比,本發明提供了一種複合發泡劑,包括磷酸鋯載體和負載於所述磷酸鋯載體上的偶氮二甲醯胺。與現有技術相比,本發明的複合發泡劑除能夠將發泡劑細化至納米級別外,其發泡劑載體對發泡劑還具有高負載量,能夠使發泡劑的負載量高達上千毫克/克,有利於降低使用成本。另外,本發明提供的製備方法簡便易行,便於實際生產應用。
附圖說明
為了更清楚地說明本發明實施例或現有技術中的技術方案,下面將對實施例或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖僅僅是本發明的實施例,對於本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據提供的附圖獲得其他的附圖。
圖1為本發明實施例1提供的複合發泡劑粉體外觀圖;
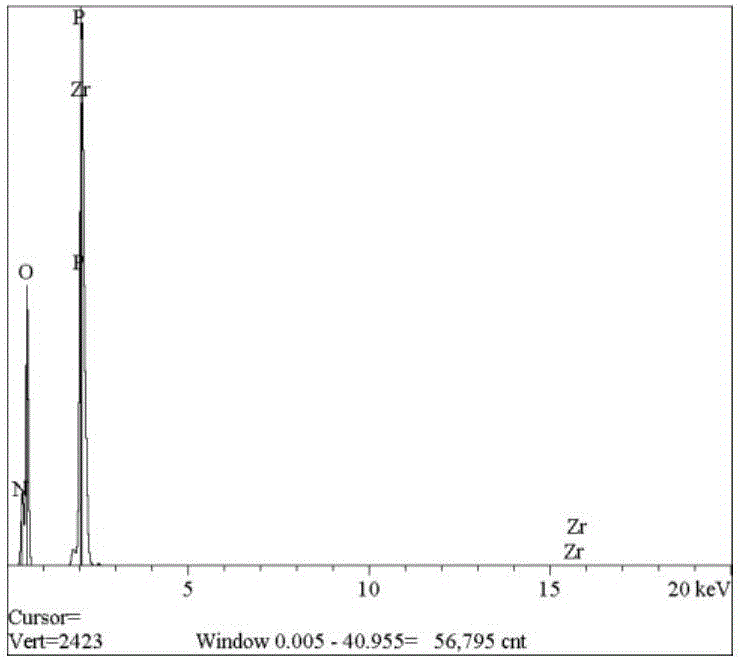
圖2為本發明實施例1提供的複合發泡劑的能譜測試圖;
圖3為本發明實施例1提供的複合發泡劑的掃描電鏡測試圖;
圖4為本發明實施例1提供的複合發泡劑的熱重分析曲線圖。
具體實施方式
本發明提供了一種複合發泡劑,包括磷酸鋯載體和負載於所述磷酸鋯載體上的偶氮二甲醯胺。
本發明中,所述磷酸鋯作為複合發泡劑中的載體來負載發泡劑,所述磷酸鋯優選為α-磷酸鋯(即α-ZrP)。本發明中,所述磷酸鋯的片層厚度優選為5~20nm;其有利於實現發泡劑的超細化,減少發泡劑的團聚。
本發明對磷酸鋯的來源沒有特殊限制,可以為市售,也可以為自製,其製備方法可以按照如下過程進行:
將氧氯化鋯(ZrOCl2·8H2O)、濃鹽酸(36wt%-38wt%)加入到去離子水中配成溶液Ⅰ;將濃磷酸(85wt%)、濃鹽酸(36wt%-38wt%)加入到去離子水中配成溶液Ⅱ。將溶液Ⅱ在室溫下連續滴加到溶液Ⅰ中,反應1小時,得到懸濁液。按照兩種溶液混合後的總體積計算,各反應物料濃度分別為:氧氯化鋯(ZrOCl2·8H2O)濃度為0.044~0.36mol/L,磷酸濃度為2.0~10mol/L,鹽酸濃度為0.50~4.0mol/L。
將所得懸濁液轉移到100ml的聚四氟乙烯容器中,裝滿度為40%-80%,將聚四氟乙烯容器密封於水熱釜中,在180~240℃下水熱處理48~192小時,將水熱釜自然冷卻至室溫,開釜抽濾,用去離子水洗滌濾餅至濾液PH值為6~7,將濾餅在烘箱中50~60℃下乾燥,得到磷酸鋯產品。
本發明的複合發泡劑中,優選的,偶氮二甲醯胺分散於磷酸鋯載體片層間和/或吸附於磷酸鋯片層表面,實現了偶氮二甲醯胺的超細化,減少發泡劑的團聚,有利於在發泡基體中實現納米尺度的均勻分散。
本發明的複合發泡劑中,所述偶氮二甲醯胺與磷酸鋯的質量比優選為(0.25~2):1,更優選為(0.29~2):1,進一步優選為(0.5~2):1,即載體對發泡劑具有優異的負載效果,其負載量可高達上千毫克/克,有利於降低使用成本。
本發明提供的複合發泡劑,不僅能夠使發泡劑細化至納米級別,克服發泡劑易團聚、難分散的缺陷,而且,所述複合發泡劑中,載體對發泡劑具有優異的負載效果,負載量可高達上千毫克/克,添加少量本發明產品即可達到良好的發泡效果,有利於降低使用成本。另外,申請人經長期研究探索發現,磷酸鋯載體與偶氮二甲醯胺的搭配,有助於降低發泡過程中氣泡成核位壘,提高成核速度,增大發泡基體的泡孔密度;且磷酸鋯片層還對氣體的逃逸有一定阻隔作用,遏制發泡過程中的氣體損失;且使用過程中,磷酸鋯的片層會在偶氮二甲醯胺分解產生的氣流作用下解離破碎,均勻分散,提高發泡基體的熔體強度,減少泡孔破裂。
本發明還提供了上述技術方案所述複合發泡劑的製備方法,包括以下步驟:
A)將偶氮二甲醯胺與溶劑混合,形成發泡劑溶液;
B)將磷酸鋯與所述發泡劑溶液混合,過濾,得到複合發泡劑。
按照本發明,首先將偶氮二甲醯胺與溶劑混合,形成發泡劑溶液。
其中,偶氮二甲醯胺為市售常規微米級產品即可。本發明中,所述溶劑優選為二甲基亞碸和/或二甲基甲醯胺。本發明對溶劑的來源沒有特殊限制,為一般市售品即可。
本發明中,所述偶氮二甲醯胺的質量與溶劑的體積比優選為(1~10)g:(10~500)mL。本發明中,偶氮二甲醯胺與溶劑混合的溫度優選為30~80℃,更優選為40~70℃;所述混合的時間優選為10~30min。
按照本發明,在得到發泡劑溶液後,將磷酸鋯與所述發泡劑溶液混合,過濾,得到複合發泡劑。
本發明中,所述磷酸鋯與上述技術方案所述一致,在此不再贅述。本發明中,混合時,發泡劑溶液中的偶氮二甲醯胺與投入的磷酸鋯的質量比優選為(0.25~2):1,更優選為(0.5~2):1,進一步優選為(0.75~2):1。
本發明中,磷酸鋯與發泡劑溶液混合的溫度優選為40~100℃,更優選為50~80℃。所述混合的時間優選為2~48小時,更優選為6~24小時,混合後,得到複合發泡劑,原本的微米級偶氮二甲醯胺發泡劑分散於磷酸鋯載體層間和/或吸附於磷酸鋯片層表面,實現了超細化分散。
本發明中,在所述混合和過濾後,優選還進行洗滌和乾燥,在所述洗滌和乾燥後,得到複合發泡劑。即將磷酸鋯與發泡劑溶液混合過濾後,對濾餅進行洗滌;所述洗滌優選為用足量的二甲基亞碸或丙酮洗滌,洗滌次數優選為兩次以上。洗滌後,再進行乾燥,即可得到複合發泡劑。所述乾燥優選為在真空環境下乾燥,乾燥的溫度優選為50~100℃。
本發明提供了一種製備上述技術方案所述複合發泡劑的方法,可以看出,本發明的製備方法簡單易行,便於實際規模化生產應用。
為了進一步理解本發明,下面結合實施例對本發明優選實施方案進行描述,但是應當理解,這些描述只是為進一步說明本發明的特徵和優點,而不是對本發明權利要求的限制。
實施例1
將2.5gAC發泡劑溶於100mL二甲基亞碸中,於60℃下恆溫攪拌混合20min,得到發泡劑溶液;向發泡劑溶液中加入2.0gα-磷酸鋯(α-ZrP),在70℃下攪拌混合15小時,得到混合溶液;將混合溶液沉澱過濾,並用二甲基亞碸對濾餅洗滌兩次後,再過濾,並置於真空烘箱中,在70℃下乾燥後,得到α-ZrP-AC納米複合發泡劑。
將所得納米複合發泡劑研磨,成為乾燥細膩的粉體,如圖1所示,所述粉體可保持長時間不結團。
對所得納米複合發泡劑分別進行能譜分析測試和掃描電鏡測試,其結果分別如圖2和圖3所示。由圖2可以看出,磷酸鋯表面N含量大幅提高,說明AC發泡劑負載於其表面;由圖3可以看出,所得複合發泡劑為層狀結構,其片層厚度為5~20nm。
對α-ZrP原料和α-ZrP-AC納米複合發泡劑進行熱重分析,測試結果如圖4所示。由圖4可以看出,從室溫加熱到500℃,α-ZrP的失重率很小,僅約為3%;而α-ZrP-AC複合發泡劑約有51%的失重率,由AC的熱分解造成。整個失重過程分為三個階段:第一失重臺階在160~225℃之間,此階段主要由AC分解產生N2、CO2、及少量的HNCO,失重率約為33%;第二失重臺階在225~250℃之間,此時第一階段反應生成的尿素發生熱分解,產生HNCO和少量的NH3,失重率約為8%;第三失重臺階在250~345℃之間,主要是聯二脲發生環化反應脫出NH3,還有少量CO2的釋放,失重率約為10%。由此可知,α-ZrP-AC納米複合發泡劑中,AC發泡劑與α-ZrP的質量比約為1:1,α-ZrP載體對AC發泡劑的負載量為1040mg/g。
實施例2
將0.5gAC發泡劑溶於100mL二甲基亞碸中,於40℃下恆溫攪拌混合20min,得到發泡劑溶液;向發泡劑溶液中加入2.0gα-磷酸鋯(α-ZrP),在70℃下攪拌混合15小時,得到混合溶液;將混合溶液沉澱過濾,並用二甲基亞碸對濾餅洗滌兩次後,再過濾,並置於真空烘箱中,在70℃下乾燥後,得到α-ZrP-AC納米複合發泡劑。
按照實施例1的測試方法對所得複合發泡劑的形貌和負載量進行測試,結果顯示,所得複合發泡劑中,AC發泡劑插入α-ZrP層間和吸附在α-ZrP層表面,細化至納米尺度,複合發泡劑的片層厚度為5~20nm。AC發泡劑與α-ZrP的質量比為0.296:1,即α-ZrP載體對AC發泡劑的負載量為296mg/g。
實施例3
將1gAC發泡劑溶於200mL二甲基甲醯胺中,於50℃下恆溫攪拌混合20min,得到發泡劑溶液;向發泡劑溶液中加入2.0gα-磷酸鋯(α-ZrP),在50℃下攪拌混合20小時,得到混合溶液;將混合溶液沉澱過濾,並用丙酮對濾餅洗滌兩次後,再過濾,並置於真空烘箱中,在80℃下乾燥後,得到α-ZrP-AC納米複合發泡劑。
按照實施例1的測試方法對所得複合發泡劑的形貌和負載量進行測試,結果顯示,所得複合發泡劑中,AC發泡劑插入α-ZrP層間和吸附在α-ZrP層表面,細化至納米尺度,複合發泡劑的片層厚度為5~20nm。AC發泡劑與α-ZrP的質量比為0.328:1,即α-ZrP載體對AC發泡劑的負載量為328mg/g。
實施例4
將1.5gAC發泡劑溶於300mL二甲基亞碸中,於80℃下恆溫攪拌混合30min,得到發泡劑溶液;向發泡劑溶液中加入2.0gα-磷酸鋯(α-ZrP),在60℃下攪拌混合24小時,得到混合溶液;將混合溶液沉澱過濾,並用二甲基亞碸對濾餅洗滌兩次後,再過濾,並置於真空烘箱中,在70℃下乾燥後,得到α-ZrP-AC納米複合發泡劑。
按照實施例1的測試方法對所得複合發泡劑的形貌和負載量進行測試,結果顯示,所得複合發泡劑中,AC發泡劑插入α-ZrP層間和吸附在α-ZrP層表面,細化至納米尺度,複合發泡劑的片層厚度為5~20nm。AC發泡劑與α-ZrP的質量比為0.563:1,即α-ZrP載體對AC發泡劑的負載量為563mg/g。
實施例5
將2.0gAC發泡劑溶於300mL二甲基亞碸中,於60℃下恆溫攪拌混合20min,得到發泡劑溶液;向發泡劑溶液中加入2.0gα-磷酸鋯(α-ZrP),在60℃下攪拌混合24小時,得到混合溶液;將混合溶液沉澱過濾,並用二甲基亞碸對濾餅洗滌兩次後,再過濾,並置於真空烘箱中,在60℃下乾燥後,得到α-ZrP-AC納米複合發泡劑。
按照實施例1的測試方法對所得複合發泡劑的形貌和負載量進行測試,結果顯示,所得複合發泡劑中,AC發泡劑插入α-ZrP層間和吸附在α-ZrP層表面,細化至納米尺度,複合發泡劑的片層厚度為5~20nm。AC發泡劑與α-ZrP的質量比為0.824:1,即α-ZrP載體對AC發泡劑的負載量為824mg/g。
實施例6
將3.0gAC發泡劑溶於300mL二甲基亞碸中,於70℃下恆溫攪拌混合20min,得到發泡劑溶液;向發泡劑溶液中加入2.0gα-磷酸鋯(α-ZrP),在80℃下攪拌混合30小時,得到混合溶液;將混合溶液沉澱過濾,並用二甲基亞碸對濾餅洗滌兩次後,再過濾,並置於真空烘箱中,在70℃下乾燥後,得到α-ZrP-AC納米複合發泡劑。
按照實施例1的測試方法對所得複合發泡劑的形貌和負載量進行測試,結果顯示,所得複合發泡劑中,AC發泡劑插入α-ZrP層間和吸附在α-ZrP層表面,細化至納米尺度,複合發泡劑的片層厚度為5~20nm。AC發泡劑與α-ZrP的質量比為1.300:1,即α-ZrP載體對AC發泡劑的負載量為1300mg/g。
實施例7
將4.0gAC發泡劑溶於200mL二甲基亞碸中,於60℃下恆溫攪拌混合20min,得到發泡劑溶液;向發泡劑溶液中加入2.0gα-磷酸鋯(α-ZrP),在70℃下攪拌混合24小時,得到混合溶液;將混合溶液沉澱過濾,並用二甲基亞碸對濾餅洗滌兩次後,再過濾,並置於真空烘箱中,在80℃下乾燥後,得到α-ZrP-AC納米複合發泡劑。
按照實施例1的測試方法對所得複合發泡劑形貌和負載量進行測試,結果顯示,所得複合發泡劑中,AC發泡劑插入α-ZrP層間和吸附在α-ZrP層表面,細化至納米尺度,複合發泡劑的片層厚度為5~20nm。AC發泡劑與α-ZrP的質量比為1.703:1,即α-ZrP載體對AC發泡劑的負載量為1703mg/g。
由以上實施例可知,本發明提供的複合發泡劑中,偶氮二甲醯胺發泡劑能夠實現超細化,以納米顆粒形式負載於載體上,而且,載體與發泡劑能夠很好的搭配,具有優異的負載效果,可使負載量高達上千毫克/克。
以上實施例的說明只是用於幫助理解本發明的方法及其核心思想。對這些實施例的多種修改對本領域的專業技術人員來說將是顯而易見的,本文中所定義的一般原理可以在不脫離本發明的精神或範圍的情況下,在其它實施例中實現。因此,本發明將不會被限制於本文所示的這些實施例,而是要符合與本文所公開的原理和新穎特點相一致的最寬的範圍。