氟硼螢光染料粘度計及其製備方法和應用與流程
2023-06-18 12:33:01 3
本發明涉及螢光染料領域,具體地,涉及氟硼螢光染料粘度計及其製備方法和應用。
背景技術:
氟硼二吡咯螢光染料(bodipy)是近二十幾年才發展起來的一類光物理化學性能優異的螢光染料分子,具有窄的吸收峰和發射峰、較高的摩爾吸光係數、較高的螢光量子產率、較好的光穩定性以及化學穩定性。普通的螢光染料若對其meso-位置的基團進行限制也會對粘度變化有一定的響應,螢光會發生變化,螢光壽命有時也會改變。
因此,製備出一種更有優異特性的螢光染料粘度計也是具有十分重要的意義。π-分子線在分子電子學和納米技術等新興領域有著獨特的優勢,含有丁二炔的分子線更加能引起人們的興趣。在這些分子線中,二聚的分子線其實是一種很好的粘度計,可以對其進行修飾,從而得到各種想要的螢光粘度計,以適應不同情況粘度的測量。
技術實現要素:
本發明的目的是提供一種氟硼螢光染料粘度計及其製備方法和應用,該氟硼螢光染料粘度計在不同粘度的溶劑內測得螢光信號不一樣,且螢光信號會隨著粘度變化呈現規律性的變化;故該氟硼螢光染料粘度計能夠作為粘度探針以及在螢光標記領域具有良好的應用前景。同時該製備方法步驟簡單,且原料易得。
為了實現上述目的,本發明提供了一種氟硼螢光染料粘度計,其中,所述氟硼螢光染料粘度計的結構如式(ⅰ)所示,
式(ⅰ)中,r1為h或c1-c3的烷基;r2為h或c1-c10的烷基;r3為h或c1-c3的烷基;x為滷素。
本發明還提供了一種上述氟硼螢光染料粘度計的製備方法,其中,所述製備方法包括如下步驟:
1)在溶劑存在條件下,將如式(a)所示的化合物與弱鹼進行第一接觸反應,製得如式(b)所示的化合物;
2)在鈀催化劑和銅催化劑的存在下,將如式(b)所示的化合物與4-碘吡啶進行第二接觸反應,製得如式(c)所示的化合物;
3)在黑暗條件下,將如式(c)所示的化合物與滷代烴進行第三接觸反應,製得如式(ⅰ)所示的化合物;
r1為h或c1-c3的烷基;r2為h或c1-c10的烷基;r3為h或c1-c3的烷基;x為滷素。
本發明還提供了一種上述氟硼螢光染料粘度計在螢光標記領域中以及作為粘度探針的應用。
通過上述技術方案,本發明通過提供的製備方法製得的氟硼螢光染料粘度計,該氟硼螢光染料粘度計在不同粘度的溶劑的螢光信號會產生規律性變化,故該氟硼螢光染料粘度計能夠作為粘度計使用;另外,該氟硼螢光染料粘度計為水溶性化合物,其可在生物細胞內進行染色分析。即該氟硼螢光染料粘度計在作為粘度探針以及螢光標記等領域具有良好的應用前景,同時其製備方法步驟簡單,產率高且原料易得。
本發明的其他特徵和優點將在隨後的具體實施方式部分予以詳細說明。
附圖說明
附圖是用來提供對本發明的進一步理解,並且構成說明書的一部分,與下面的具體實施方式一起用於解釋本發明,但並不構成對本發明的限制。在附圖中:
圖1是檢測例2中的螢光發射光譜圖;
圖2是檢測例3中的螢光發射光譜圖。
具體實施方式
以下對本發明的具體實施方式進行詳細說明。應當理解的是,此處所描述的具體實施方式僅用於說明和解釋本發明,並不用於限制本發明。
本發明中提供了一種氟硼螢光染料粘度計,其中,所述氟硼螢光染料粘度計的結構如式(ⅰ)所示,
式(ⅰ)中,r1為h或c1-c3的烷基;r2為h或c1-c10的烷基;r3為h或c1-c3的烷基;x為滷素。
本發明中,所述r1、r2和r3均可以在寬的範圍內選擇,但是為了進一步提高所述氟硼螢光染料粘度計的水溶性及其產率,優選地,所述r1為甲基;所述r2為正庚基;所述r3為甲基,所述滷素為碘。
本發明還提供了一種上述氟硼螢光染料粘度計製備方法,其特徵在於,所述製備方法包括如下步驟:
1)在溶劑存在條件下,將如式(a)所示的化合物與弱鹼進行第一接觸反應,製得如式(b)所示的化合物;
2)在鈀催化劑和銅催化劑的存在下,將如式(b)所示的化合物與4-碘吡啶進行第二接觸反應,製得如式(c)所示的化合物;
3)在黑暗條件下,將如式(c)所示的化合物與滷代烴進行第三接觸反應,製得如式(ⅰ)所示的化合物;
其中,r1為h或c1-c3的烷基;r2為h或c1-c10的烷基;r3為h或c1-c3的烷基;x為滷素。
在上述製備方法中,所述r1、r2和r3均可以在寬的範圍內選擇,但是為了進一步提高所述氟硼螢光染料粘度計的水溶性及其產率,優選地,所述r1為甲基;所述r2為正庚基;所述r3為甲基,所述滷素為碘。
另外,步驟1)和2)是在溶劑存在下發生反應的,且溶劑種類可以在寬的範圍內選擇,但是為了促進反應的進行及提高產物的產率,優選地,所述溶劑由溶劑a和溶劑b組成;其中,溶劑a為四氫呋喃,溶劑b為甲醇或乙醇,且溶劑a與溶劑b的體積比為1:1-1.5。
為了促進步驟1)的反應進行,進一步優選地,第二接觸反應於四氫呋喃中進行。
為了促進步驟2)的反應進行,進一步優選地,第三接觸反應於n,n-二甲基甲醯胺中進行。
為了促進步驟3)的反應進行,第三接觸反應於n,n-二甲基甲醯胺中進行。
在發明提供的製備方法中,各原料的用量可以根據實際需要進行調節,在發明的一種優選的實施方式中,為了使產品的轉化率較高,進一步降低製備成本,優選地,相對於1mol所述式(a)所示的化合物,所述弱鹼的用量為4-15mol,所述4-碘吡啶的用量為2-12mol,所述滷代烴的用量為40-200mol,所述鈀催化劑的用量為0.03-0.2mol,所述銅催化劑的用量為0.03-0.2mol。
更為優選地實施方式中,相對於1mol如所述式(a)所示的化合物,所述弱鹼的用量為4-8mol,所述4-碘吡啶的用量為2-8mol,所述滷代烴的用量為60-120mol。
所述弱鹼可在寬的範圍內選擇,但是為了進一步提高產品的轉化率,降低製備成本,優選地,所述弱鹼為碳酸鉀、碳酸鈉、碳酸氫鈉或碳酸氫鉀
同時,所述滷代烴的種類可以在寬的範圍內選擇,但是為了提高反應產率,優選地,所述滷代烴為碘甲烷、溴甲烷或溴丁烷。
另外,本發明中,鈀催化劑和銅催化劑的種類可以在寬的範圍內選擇,但是為了提高反應速率和產品的轉換率,優選地,所述鈀催化劑為二氯二三苯基磷鈀,所述銅催化劑為氯化亞銅。
此外,在本發明中另一種優選地實施方式中使得經過第二接觸反應製得的氟硼螢光染料粘度計優異的產率,第二接觸反應體系中還包括路易斯鹼,且相對於1mol所述式(a)所示的化合物,所述路易斯鹼的用量為20-80mol;更優選地,所述路易斯鹼的用量為40-60mol。
其中,路易斯鹼的具體種類可以在寬的範圍內選擇,但是為了進一步地提高產率以及從成本上考慮,優選地,所述路易斯鹼為三乙胺。
第一接觸反應、第二接觸反應和第三接觸反應的反應條件可以不作進一步限定,當然,在本發明的一種優選的實施方式中,為了使原料的轉化率更高,優選地,所述第一接觸反應的反應時間為0.5-3h,反應溫度為15-40℃;所述第二接觸反應的反應時間為0.5-3h,反應溫度為20-60℃;所述第三接觸反應的反應時間為6-15h,反應溫度為25-40℃。
本發明還提供了上述氟硼螢光染料粘度計以及根據上述的方法製備的氟硼螢光染料粘度計在螢光標記以及作為粘度探針等領域中的應用。
以下將通過實施例對本發明進行詳細描述,但本發明並不僅限於下述實施例。
以下實施例中,核磁測定採用瑞士bruker公司的av-300型核磁共振儀進行;質譜的測定採用美國儀器集團的hplc/esi-ms型質譜儀進行;紫外光譜的測定採用日本島津公司的uv-2450型紫外/可見分光光度計進行,螢光光譜的測定日本日立公司的f-4500fl螢光分光光度計進行,相對螢光量子產率的測定採用螢光光譜的測定日本日立公司的f-4500fl螢光分光光度計進行,單晶衍射的測定採用德國brukeraxs公司的smarapexⅱx-單晶衍射儀進行,其中λmax表示最大吸收波長、εabs表示摩爾消光係數,λemmax表示最大螢光發射波長,φf表示相對螢光量子產率和stokes-shift表示stokes位移;相對螢光量子產率(φf)的測定是以其中相對螢光量子產率φf的測定以螢光黃(φ=0.90,在氫氧化鈉溶液中)為標準染料,根據公式φf=φs*(ix/is)*(as/ax)*(nx/ns)2計算所得,其中φs為標準物螢光黃的螢光量子產率,i為譜圖積分面積,a為吸光度,n為溶劑的折光率,下角標s為標準物,x為待測物。
以下實施例中使用的原料:碳酸鉀、甲醇、四氫呋喃、己烷、三乙胺、二氯甲烷、n,n-二甲基甲醯胺是國藥集團化學試劑有限公司的產品,碘化亞銅、二氯二三苯基磷化鈀、4-碘吡啶、碘甲烷是安耐吉化學公司的產品。
製備例1
將如式(3b-1)所示結構的原料(108mg,0.2mmol),cucl(160mg,1.6mmol)加入到小試管內,然後加入1ml乾燥的n,n-二甲基甲醯胺,放在60℃下反應2小時。將反應液倒入水中,用二氯甲烷萃取,用無水硫酸鈉乾燥後,減壓蒸餾,而後再用矽膠柱層析製得如式(a』)所示結構的化合物(摩爾產率為37%),有7%的原料未反應完。
對上述如式(a』)所示結構的化合物進行核磁氫譜和核磁碳譜檢測:
1hnmr(500mhz,cdcl3)δ:2.98(t,j=7.8hz,4h),2.65(s,6h),2.61(s,6h),2.54(s,6h),2.52(s,6h),1.66-1.58(m,4h),1.52-1.49(m,4h),1.37-1.27(m,12h),0.90(t,j=6.8hz,6h),0.26(s,18h);13cnmr(75mhz,cdcl3)δ:158.0,157.6,148.2,142.7,142.5,131.5,130.9,116.8,114.7,102.1,97.1,80.7,75.6,31.8,31.7,30.2,29.0,28.8,22.6,15.1,14.1,14.0,13.7,13.5;hrms(apci)calcd.forc54h73b2f4n4si2[m+h]+:931.5491,found:931.5451。
製備例2
如式(b』)所示結構的原料的製備:將如式(a』)所示結構的原料(93mg,0.1mmol)溶解在6ml的甲醇和6ml的四氫呋喃裡,然後加入碳酸鉀(83mg,0.6mmol),在20℃條件下攪拌1小時。反應完全後將反應液倒入稀鹽酸水溶液中,用二氯甲烷萃取,用無水硫酸鈉乾燥後,減壓蒸餾,而後再用矽膠柱層析製得如式(b』)所示結構的化合物(產率為92%)。
對上述如式(b』)所示結構的化合物進行核磁氫譜和核磁碳譜檢測:1hnmr(500mhz,cdcl3)δ:3.40(s,2h),2.99(t,j=8.0hz,4h),2.66(s,6h),2.63(s,6h),2.55(s,6h),2.53(s,6h),1.67-1.59(m,4h),1.53-1.51(m,4h),1.37-1.30(m,12h),0.90(t,j=6.5hz,6h);13cnmr(125mhz,cdcl3)δ:158.0,157.8,148.4,143.1,142.9,131.4,131.0,115.5,114.8,84.4,80.6,76.0,75.5,31.8,31.7,30.3,29.0,28.9,22.6,15.3,15.1,14.1,13.7,13.5。
製備例3
如式(c』)所示結構的原料的製備:將如式(b』)所示結構的原料(79mg,0.1mmol),4-碘吡啶(123mg,0.6mmol),pd(pph3)2cl2(7mg,0.01mmol)和cui(2mg,0.01mmol)加入到schlenk瓶內,抽真空通氬氣三次,在通氬氣氛圍下將5ml四氫呋喃和0.5ml三乙胺加入瓶內,在40℃氬氣氛圍下反應1小時,反應完全後將反應液經過一個短矽膠柱,除去不溶的催化劑,減壓蒸餾,而後再用矽膠柱層析製得如式(c』)所示結構的的化合物(產率為81%)。
對上述如式(c』)所示結構的的化合物進行核磁氫譜和核磁碳譜檢測:1hnmr(500mhz,cdcl3)δ:8.61(d,j=5.0hz,4h),7.38(d,j=5.0hz,4h),3.02(t,j=8.3hz,4h),2.69(s,6h),2.68(s,6h),2.59(s,6h),2.57(s,6h),1.85-1.75(m,4h),1.64-1.63(m,4h),1.38-1.29(m,12h),0.91(t,j=7.5hz,6h);13cnmr(125mhz,cdcl3)δ:158.6,157.3,149.8,148.6,143.4,142.3,131.6,131.5,131.3,125.1,115.2,94.1,86.7,80.9,75.5,31.9,31.7,30.3,29.1,29.0,22.6,15.3,15.2,14.1,13.8,13.7。
實施例1
如式(i』)所示的氟硼螢光染料粘度計的製備:將如式(c』)所示結構的化合物(19mg,0.02mmol)加入到小試管內,然後加入1mln,n-二甲基甲醯胺,再加入碘甲烷(125μl,2mmol),放在30℃黑暗條件下反應10小時。反應完成後在反應液上面鋪一層乙醚將產物重結晶沉澱出來,然後進行離心,倒掉上清液即可得到如式(i』)所示的氟硼螢光染料粘度計(產率為91%)。
對上述如式(i』)所示的氟硼螢光染料粘度計進行核磁氫譜、核磁碳譜檢測:1hnmr(500mhz,dmso-d6)δ:8.97(d,j=7.0hz,4h),8.23(d,j=7.0hz,4h),4.29(s,6h),3.09(t,j=5.0hz,4h),2.66-2.65(m,12h),2.60-2.58(m,12h),1.65-1.63(m,4h),1.55-1.53(m,4h),1.40-1.38(m,4h),1.32-1.30(m,8h),0.89(t,j=6.5hz,6h);13cnmr(125mhz,dmso-d6)δ:159.2,157.4,150.7,145.7,144.4,138.8,132.0,131.7,131.5,128.6,115.1,113.5,96.2,93.8,81.1,76.0,48.0,31.6,31.4,30.0,28.9,28.7,22.5,15.6,15.4,14.3,14.0,13.8。
實施例2
按照實施例1的方法:將如式(c』)所示結構的化合物(19mg,0.02mmol)加入到小試管內,然後加入1mln,n-二甲基甲醯胺,再加入碘甲烷(125μl,2mmol),放在35℃黑暗條件下反應12小時。反應完成後在反應液上面鋪一層乙醚將產物重結晶沉澱出來,然後進行離心,倒掉上清液即可得到如式(i』)所示的氟硼螢光染料粘度計(產率為91%)。
實施例3
按照實施例1的方法:將如式(c』)所示結構的化合物(19mg,0.02mmol)加入到小試管內,然後加入1mln,n-二甲基甲醯胺,再加入碘甲烷(125μl,2mmol),放在40℃黑暗條件下反應15小時。反應完成後在反應液上面鋪一層乙醚將產物重結晶沉澱出來,然後進行離心,倒掉上清液即可得到如式(i』)所示的氟硼螢光染料粘度計(產率為91%)。
測試例1
將上述製備例1-3製得的原料(a』)、(b』)、(c』)和實施例1中製得的氟硼螢光染料粘度計(i』)分別檢測其在不同溶劑中的光譜性質,測試結果如表1所示:
表1
表1中:stokes-shift=λemmax-λmax(nm)。
檢測例2
將實施例1中製備的如式(i』)所示結構的粘度計在乙醇和丙三醇不同體積比溶劑中的螢光發射光譜圖,結構見圖1所示;其中圖1中所標數字為乙醇、丙三醇不同比例下所對應的溶液的粘度數值。
圖1中,隨著粘度的增加,氟硼螢光染料粘度計中兩個氟硼二吡咯單元繞中間的丁二炔旋轉速度變慢,其共平面性在減弱,相互扭曲的構象在增加。所得螢光光譜中長波峰的強度在降低,短波峰的強度在增加。
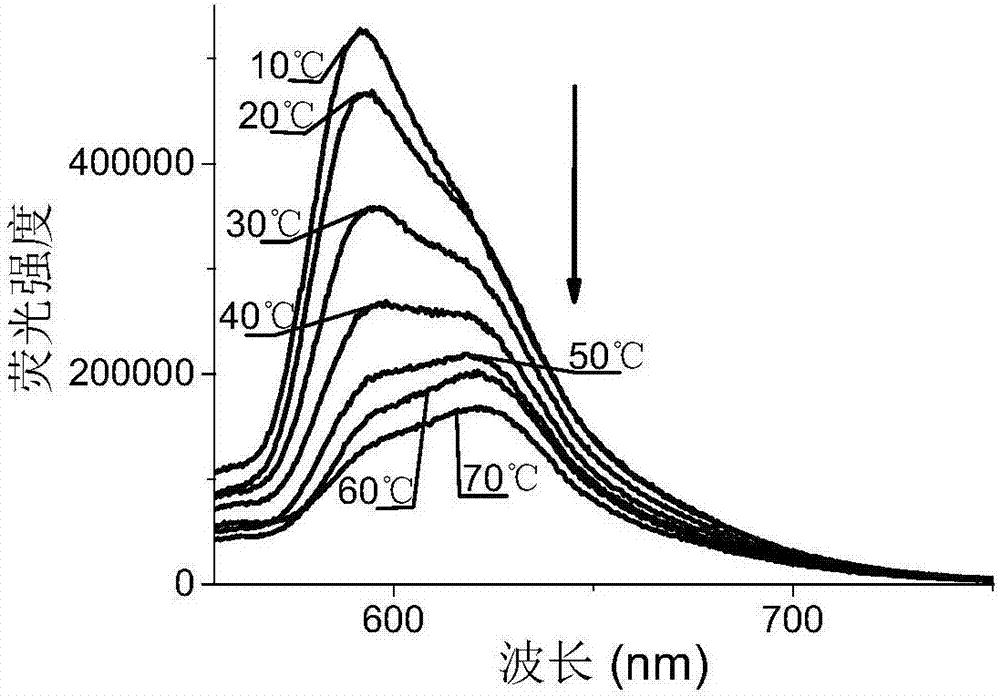
檢測例3
將實施例1中製備的如式(i』)所示結構的氟硼螢光染料粘度計在丙三醇與乙醇的體積比為9:1的混合溶劑中,測試不同溫度的螢光發射光譜圖,結果見圖2所示,圖2中所標數值為測時溶劑的溫度數值。
圖2中,隨著溫度的升高,混合溶劑的粘度數值在降低,此時測試的螢光光譜也發生改變,進一步說明本發明提供的氟硼螢光染料粘度計在測量粘度變化方面具有良好的應用價值。
通過上述圖1和圖2可知,本發明製得的氟硼螢光染料粘度計在乙醇裡的最大螢光發射波長在624nm,說明其兩邊的氟硼二吡咯單元在低粘度介質中處於共平面位置,共軛比較好,螢光發射波長比較長;當增加介質的粘度時,粘度計中兩個氟硼二吡咯單元繞中間的丁二炔旋轉的速度變慢,染料分子的平面性受到破壞,共軛效果降低,導致螢光發射波長藍移。同時該氟硼螢光染料粘度計還具有水溶性,其可在生物細胞內進行染色分析。說明其在螢光標記以及作為粘度探針等領域具有良好的應用前景,同時該製備方法步驟簡單,產率高且原料易得。
同樣,對上述實施例2-3所製得的如式(i』)所示的氟硼螢光染料粘度計進行核磁氫譜、核磁碳譜檢測,其結果與實施例1的檢測結果基本一致;並對實施裡2-3製得的如式(i』)所示的氟硼螢光染料粘度計進行檢測例1-3的檢測,其檢測結果也與實施例1的檢測結果基本一致。
所示實施例僅用於描述本發明的概要,並不限制本發明,技術員可在所屬領域自主選擇實施。
以上詳細描述了本發明的優選實施方式,但是,本發明並不限於上述實施方式中的具體細節,在本發明的技術構思範圍內,可以對本發明的技術方案進行多種簡單變型,這些簡單變型均屬於本發明的保護範圍。
另外需要說明的是,在上述具體實施方式中所描述的各個具體技術特徵,在不矛盾的情況下,可以通過任何合適的方式進行組合,為了避免不必要的重複,本發明對各種可能的組合方式不再另行說明。
此外,本發明的各種不同的實施方式之間也可以進行任意組合,只要其不違背本發明的思想,其同樣應當視為本發明所公開的內容。