靶向人TSPAN8基因的siRNA及其應用的製作方法
2023-06-04 11:15:51 1
本發明涉及生物醫藥及基因治療領域,具體涉及一種靶向人TSPAN8基因的siRNA及其在製備抗腫瘤藥物中的應用。
背景技術:
TSPAN8的英文全稱是tetraspanin 8(四跨膜蛋白8),又稱為CO-029,TM4SF3,位於12號染色體上(Genebank Accession:NM_004616)。TSPAN8是四跨膜超家族重要成員之一。TSPAN8通過影響吞噬細胞糖蛋白1(phagocyticglycoprotein 1,CD44)(Guo Q.ect,Tetrasoanin CO-029inhibit colorectal cancer cell movement by deregulationg cell-matrix and cell-cell adhensions,PloS One.2012;7(6)e:38464),或是影響局部粘著斑激酶(Focal Adhesion Kinase,FAK),或是影響樁蛋白(Paxilin)(Shijing Yue等,Tspan8and CD151promote metastasis by distinct mechanisms.European Journal of Cancer(2013)49,2934–2948),進而影響層粘連蛋白整合素(laminin-binding intergrinα3β1)和成纖維粘連蛋白整合素(fibronectin-intergrinα5β1)的表達,降低細胞的遷移和黏附能力。TSPAN8在細胞表面與整合素、生長因子受體以及其他四跨膜超蛋白形成四跨膜網絡,並對整合素、生長因子受體的功能產生影響。目前研究表面其參與細胞的黏附、遷移及增殖等眾多病理生理過程。(Wu Jin-cai等,The influence of down-regulation of Tspan8by shRNA on metastasis and invasion of hepatocellular carcinomas,China J Hepatobiliary Surg,February 2012,Vol.18,No.2)。
腫瘤是威脅人類健康的疾病之一,至今缺少理想的治療手段和藥物,開 發新的藥物迫在眉睫。基因治療是非常有前景的,在不久的將來或許會成為腫瘤治療的一個有效手段。RNA幹擾技術是一種沉默基因表達的技術,兩名美國科學家Andrew Z.Fire和Craig C.Mello因對此項技術的貢獻於2006年被授予諾貝爾生理學獎。RNA幹擾技術的原理是較長雙鏈RNA被特異核酸酶Dicer切割加工成21-23nt的由正義和反義鏈組成的小幹擾RNA。小幹擾RNA隨後形成沉默複合體(RNA-induced silencing complex,RISC)解旋成單鏈。反義鏈引導該沉默複合體通過鹼基配對特異性結合到目標mRNA上,使mRNA分解。
小髮夾RNA(short hairpin RNA,shRNA)是一種形成急轉彎結構的RNA序列,可以經由RNA幹擾使基因沉默。
膠質瘤是一種發生於腦或脊髓的腫瘤,最常發生部位為腦,因其來源神經膠質細胞而被稱為膠質瘤。膠質瘤佔腦和中樞神經系統腫瘤的30%、佔腦惡性腦腫瘤的80%,是人類健康的嚴重威脅。腦膠質瘤的治療通常採用手術,放療和化療相結合。膠質瘤常發生於腦,手術需要開顱,手術時間長。膠質瘤和正常神經組織交錯生長,邊界不清,瘤組織不易清理乾淨,膠質瘤易復發。對於化療,由於血腦屏障的存在,使普通的抗腫瘤藥物療效不佳。對於放療,也存在定位困難和對神經損傷的擔心。目前膠質瘤仍是醫學界的一個難題。
當前有關TSPAN8的研究主要集中在肝癌消化道癌領域,在膠質瘤基因治療領域還是一片空白。
技術實現要素:
本發明的目的在於提供了一種靶向人TSPAN8基因的siRNA,本發明的另一目的在於提供該siRNA在製備抗腫瘤藥物中的應用。
本發明人經實驗證明惡性程度較高的膠質瘤,TSPAN8的表達量也較高,調節TSPAN8的表達能影響膠質瘤細胞的增殖。
本發明結合TSPAN8分子在瘤細胞中的功能,利用RNA幹擾技術開發新的治療藥物。
本發明設計靶向TSPAN8基因的siRNA,特異性抑制TSPAN8基因的表達,從而抑制膠質瘤的增殖。進一步地本發明提供了包含該siRNA的shRNA分子,也具有抑制TSPAN8的表達的作用,可能成為治療膠質瘤的藥物。
本發明的第一方面,是提供一種靶向人TSPAN8基因,並特異性抑制TSPAN8基因表達的siRNA或shRNA。
本發明提供了一種靶向人TSPAN8基因並特異性抑制TSPAN8基因表達的siRNA,包括正義鏈和反義鏈,siRNA的序列如下:
正義鏈:5』-GCAAUGACUCUCAAGCAA-3』 (SEQ ID NO.1)
反義鏈:5』-CGUUACUGAGAGUUCGUU-3』 (SEQ ID NO.2)
本發明提供了一種包含上述的siRNA序列的shRNA序列,包括正義鏈和反義鏈,此shRNA序列為:
正義鏈:
5』-CCGGGCAAUGACUCUCAAGCAACUCGAGUUGCUUGAGAGUCAUUGC-3』(SEQ ID NO.3)
反義鏈:
5』-AAUUCAAAAAAGCAAUGACUCUCAAGCAACUCGAGUUGCUUGAGAGUCAUUGC-3』(SEQ ID NO.4)
進一步地,本發明提供了一種轉錄上述的shRNA的DNA序列,DNA序列為:
轉錄正義鏈DNA:5』-CCGGGCAATGACTCTCAAGCAACTCGAGTTGCTTGAGAGTCATTGC-3』(SEQ ID NO.5)
轉錄反義鏈DNA:5』- AATTCAAAAAAGCAATGACTCTCAAGCAACTCGAGTTGCTTGAGAGTCATTGC-3』(SEQ ID NO.6)
本發明的第二方面,提供了一種重組載體,含有如上所述的靶向人TSPAN8基因的siRNA或shRNA。
所述的重組載體,載體可選自質粒、慢病毒載體、腺病毒載體等,優選為質粒pLKD-CMV-GFP-U6。
本發明的第三方面,是提供上述的siRNA、shRNA,或DNA序列在製備TSPAN8基因表達抑制劑中的應用。
進一步地,本發明還提供了上述的siRNA、shRNA,或DNA序列在製備抗腫瘤藥物中的應用。
所述的腫瘤為膠質瘤。
本發明主要技術方案是:
1.siRNA的設計和幹擾載體的製備;
2.幹擾慢病毒顆粒的包裝;
3.幹擾效率在蛋白水平的驗證;
4.MTT法檢測幹擾TSPAN8後細胞系增殖的影響;
5.克隆形成檢測幹擾TSPAN8後對細胞系增殖的影響。
本發明涉及的幹擾RNA能在信使RNA水平和蛋白水平有效抑制人TSPAN8基因的表達,從而抑制膠質瘤細胞系U87和U251的增殖,為膠質瘤的治療提供了新的途徑。
附圖說明
圖1採用Western blot檢測TSPAN8幹擾慢病毒感染U87細胞,對目的基因蛋白表達的敲減效果
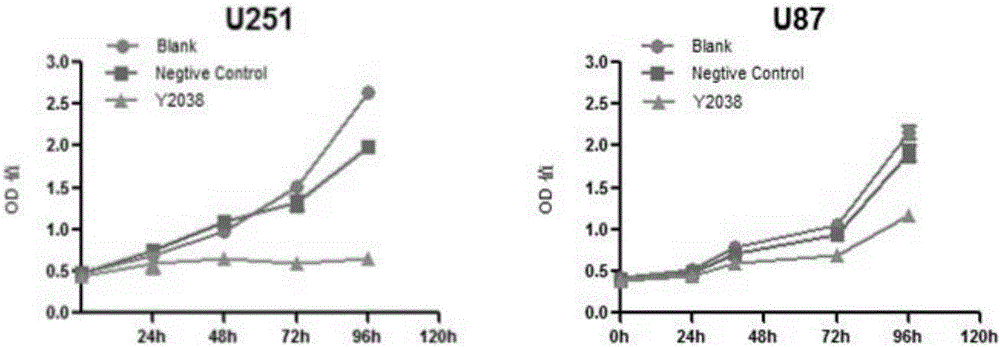
圖2為MTT法檢測幹擾TSPAN8抑制U87和U251細胞系生長的折線圖;
圖3為Transwell法檢測TSPAN8抑制U87和U251細胞侵襲的照片圖;
圖4為克隆形成法檢測幹擾TSPAN8抑制U87和U251形成克隆的照片圖。
圖2-4中含有空載體的病毒(Negative control)和幹擾TSPAN8的病毒(sh TSPAN8),Blank是指沒有經過病毒處理的細胞。
具體實施方式
下面結合附圖對本發明進一步說明。以下實施方式只是較佳的實施方式中的一種,並非對本發明的限制。其他的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護範圍之內。未作特殊說明的實驗試劑和方法,均指常規試劑和方法。
本發明所用試劑和原料均市售可得或可按文獻方法製備。下列實施例中未註明具體條件的實驗方法,通常按照常規條件如Sambrook等人《分子克隆:實驗室指南》(New York:Cold Spring Harbor Laboratory Press,1989)中所述的條件,或按照常規條件,或按照製造廠商所建議的條件。
實施例1TSPAN8siRNA序列設計與幹擾質粒構建
在ensembl網站(http://www.ensembl.org/index.html)搜索並下載TSPAN8的cDNA序列,使用Life technologies公司的BLOCK-iT RNA Designer軟體(http://rnaidesigner.lifetechnologies.com/rnaexpress/)在線設計與TSPAN8cDNA互補的siRNA序列肆條。選取得分最高的siRNA序列,如下:
siRNA1~4的序列如下:
siRNA1
正義鏈:5』-GCAAUAUGGGUACGAGUAA-3』 (SEQ ID NO.7)
反義鏈:5』-UUACUCGUACCCAUAUUGC-3』 (SEQ ID NO.8)
siRNA2
正義鏈:5』-GCAAUGACUCUCAAGCAA-3』 (SEQ ID NO.1)
反義鏈:5』-CGUUACUGAGAGUUCGUU-3』 (SEQ ID NO.2)
siRNA3
正義鏈:5』-GGAAGCCAUAAUUGUGUUU-3』 (SEQ ID NO.9)
反義鏈:5』-AAACACAAUUAUGGCUUCC-3』 (SEQ ID NO.10)
siRNA4
正義鏈:5』-GAGUUUAAAUGCUGCGGUU-3』 (SEQ ID NO.11)
反義鏈:5』-AACCGCAGCAUUUAAACUC-3』 (SEQ ID NO.12)
輸入到NCBI網站的Standard Nuceotide BLAST界面,與人的轉錄序列比對,確認目的基因與除TSPAN8以外的轉錄本無高度同源性(16個鹼基以上),排除偏靶效應。然後按照「AgeI酶切位點粘性末端-正義鏈-莖環-反義鏈-編碼終止序列-EcoRI酶切位點粘性末端」的順序,將siRNA1~4組裝入shRNA1~4(Y2037,Y2038,Y2039,Y2040)正義鏈和反義鏈。
shRNA1(Y2037):
轉錄正義鏈DNA:5』-CCGGGCAATATGGGTACGAGTAATTCAAGAGATTACTCGTACCCATATTGCTTTTTTG-3』 (SEQ ID NO.13)
轉錄正義鏈DNA:5』-AATTCAAAAAAGCAATATGGGTACGAGTAATCTCTTGAATTACTCGTACCCATATTGC-3』 (SEQ ID NO.14)
shRNA2(Y2038):
轉錄正義鏈DNA:5』-CCGGGCAATGACTCTCAAGCAACTCGAGTTGCTTGAGAGTCATTGC-3』(SEQ ID NO.5)
反義鏈DNA:5』-AATTCAAAAAAGCAATGACTCTCAAGCAACTCGAGTTGCTTGAGAGTCATTGC-3』(SEQ ID NO.6)
shRNA3(Y2039):
轉錄正義鏈DNA:5』-CCGGGGAAGCCATAATTGTGTTTCTCAAGAGAAAACACAAT TATGGCTTCCTTTTTTG-3』(SEQ ID NO.15)
轉錄正義鏈DNA:5』-AATTCAAAAAAGGAAGCCATAATTGTGTTTTCTCTTGAGAAACACAATTATGGCTTCC-3』(SEQ ID NO.16)
shRNA4(Y2040):
轉錄正義鏈DNA:5』-CCGGGAGTTTAAATGCTGCGGTTCTCAAGAGAAACCGCAGCATTTAAACTCTTTTTTG-3』(SEQ ID NO.17)
轉錄正義鏈DNA:5』-AATTCAAAAAAGAGTTTAAATGCTGCGGTTTCTCTTGA GAACCGCAGCATTTAAACTC-3』(SEQ ID NO.18)
把以上信息交invitrogen公司合成得到DNAOligo。
DNA Oligo加入ddH2O溶解,濃度10mM。各別取22.5μl的DNA Oligo與5μl的10×退火緩衝液(成分為100mMTris-HCl(pH 7.5),10mM EDTA,1mM NaCl)混合,95℃水浴12min後拿出,在室溫下冷卻至室溫。
25℃下,用T4連接酶(日本TAKARA公司生產)將互補雙鏈與AgeI、EcoRI酶切的載體連接30min,體系按說明書配製(DNA 4μl,載體2μl,PEG40002μl,10×T4buffer 2μl,T4連接酶1μl,ddH2O 9μl)。取連接產物加入裝有大腸桿菌DH5α感受態細胞的1.5ml規格離心管使其混合,置於冰上30分鐘,然後放入42℃水浴90秒,再放回冰上2分鐘後加入800μl不含抗生素的培養基,置37℃細菌培養箱中搖1小時。4500轉/分離心收集得到的細菌後塗固體LB培養基平板,37℃細菌培養箱過夜培養至長出菌落。
挑取菌落溶於含20μl抗性培養基的離心管中,進行菌落PCR鑑定,鑑定所用的引物為:
PLKD-F:CCTATTTCCCATGATTCCTTCATA(SEQ ID NO.19)
PLKD-R:GAAATACGGTTATCCACGCG(SEQ ID NO.20)
轉化成功的取樣送測序(華大基因有限公司提供測序服務),測序使用的引物是:PLKD-F:CCTATTTCCCATGATTCCTTCATA(SEQ ID NO.21)。測序結果比對正確後,應將菌液加入含15ml氨苄青黴素LB培養基的50ml離心管中。將離心管放入37℃搖床培養20小時。用質粒小提中量試劑盒(天根公 司)抽取質粒,測定DNA濃度後備用。
實施例2:幹擾慢病毒顆粒的包裝
在37℃5%CO2的細胞培養箱中培養HEK 293T細胞(下簡稱293T,購自中國科學院典型培養物保藏委員會細胞庫),培養基使用添加了10%胎牛血清(美國紐約Gibco公司生產)的DMEM(美國紐約Gibco公司生產)培養基.傳代培養293T細胞於直徑為10cm的培養皿中,當細胞長到匯合度為50%左右,用無血清的培養基培養4小時。
按Lipofectamine 2000(美國Invitrogen公司生產)說明書操作,準備好22.5g上面得到的pLKD-CMV-GFP-U6-shRNA質粒(或不含shRNA的空載體質粒)、7.9g病毒外殼質粒psPAX2(中國上海紐恩生物科技有限公司提供)、14.6g包裝質粒pMD2.G(中國上海紐恩生物科技有限公司提供)的轉染混合物。
把混合物添加到已經飢餓培養過的細胞中進行轉染,4-6小時後更換為正常的培養基。分別在換液培養24和48小時後收集培養基,經0.22μm孔徑的膜過濾後,再用CP 80MX離心機(日本日立公司生產)經4℃、100000g超速離心2小時,得到病毒沉積塊。用OPTI-MEM(美國紐約Gibco公司生產)培養基重懸收集得到慢病毒。
通過以上方法包裝得到含有空載體的病毒(control)和幹擾TSPAN8的病毒(shTSPAN8:Y2037,Y2038,Y2039,Y2040)。慢病毒的滴度測定採用稀釋計數法,把293T細胞鋪96孔板,每孔約5000個細胞,過夜培養。準備好用培養基10倍梯度稀釋的病毒共10份,即最低、最高濃度分別為原液的1/10-10、1/10-1,每份100μl換掉對應孔的培養基培養過夜後更換正常培養基。換掉新鮮培養基再培養兩天後,置螢光顯微鏡下觀察最後兩個有螢光的孔的螢光細胞的克隆數,用以下公式計算病毒滴度:滴度(TU/ml)=(X+Y*10)*10/(2*Z)其中X是指倒數第二個有螢光的孔的螢光細胞克隆 數,Y是指倒數第一個有螢光的孔的螢光細胞克隆數,Z是指X對應孔所加病毒的稀釋率。
實施例3:幹擾效率在蛋白水平的驗證
蛋白樣品的製備:
在直徑6cm培養皿中培養細胞至大約30%匯合度,分別加入對照慢病毒感染顆粒和幹擾TSPAN8的病毒感染顆粒(感染複數為10),繼續培養細胞4天。實驗分為5組,分別為:陰性對照病毒感染組(Control),Y2037病毒感染組,Y2038病毒感染組,Y2039病毒感染組和Y2040病毒感染組。收細胞時先轉移培養基至空的15ml規格離心管,用PBS漂洗兩次細胞,每次用PBS 3ml,然後用2ml 0.25%胰蛋白酶消化細胞,待細胞都變圓漂浮後把之前吸走的培養基倒回培養皿。把消化下來的細胞隨培養基轉移到15ml規格離心管中,1200轉/分離心5分鐘收集細胞。棄掉上清,加入含有1×PMSF(中國南通碧雲天公司生產)和1×蛋白酶抑制劑cocktail(美國Thermo Scientific公司生產)的RIPA細胞裂解液(中國南通碧雲天公司生產)。吹打混勻細胞和裂解液,放於冰上裂解1小時,每20分鐘用漩渦混合器振蕩混勻。最後離心,離心機設置為:4℃、13000轉/分、15分鐘。吸取離心後的上清液至潔淨的離心管中即為提得的蛋白樣品。把蛋白樣品和等體積的2×蛋白上樣緩衝液(二硫蘇糖醇(DTT):0.1572g,溴酚藍;0.01g,Tris-HCl(1M pH 6.8):0.5ml,10%SDS:2ml,甘油:1ml,H2O:定容至10ml)混合煮沸5分鐘,完成樣品製備。
Tris(1M pH 6.8)緩衝液的配製方法:稱取Tris Base 24.228g溶於約160ml超純水,充分攪拌溶解,用鹽酸調節pH至6.8,同時用超純水定容至200ml。
Western Blot方法檢測蛋白表達:
準備好配製凝膠需要的試劑:
30%acrylamide mix的配製方法:稱取丙烯醯胺29.2g,甲叉雙丙烯醯胺0.8g,用超純水溶解並定容至100ml。
Tris(1.5M pH 8.8)配製方法:稱取Tris Base 36.342g溶於約160ml超純水,充分攪拌溶解,用鹽酸調節pH至8.8,同時用超純水定容至200ml。
Tris(0.5M pH 6.8)緩衝液的配製方法:稱取Tris Base 12.114g溶於約160ml超純水,充分攪拌溶解,用鹽酸調節pH至6.8,同時用超純水定容至200ml。
10%SDS的配製方法:稱取5g十二烷基硫酸鈉(sodium dodecyl sulfate,SDS),溶於超純水至50ml。
10%APS的配製方法:稱取5g過硫酸銨(Ammonium persulfate,APS),溶於超純水至50ml。
TEMED是指N,N,N',N'-Tetramethylethylenediamine,中文名N,N,N',N'-四甲基二乙胺。
準備好電泳裝置(中國上海天能科技有限公司生產的VE-180型垂直電泳槽)和配製1.5mm厚凝膠的玻璃板(中國上海天能科技有限公司生產)。把玻璃板用洗潔精、清水洗滌乾淨,再用去離子水衝洗一遍,放入電熱鼓風乾燥箱中烘乾。待玻璃板全乾後組裝好配膠裝置,配製10%分離膠(ddH2O:4.0ml,30%acrylamide mix:3.3ml,Tris(PH 8.8 1.5M):2.5ml,10%SDS:100μl,10%APS:100μl,TEMED:4μl),將其注入至玻璃板,加入後用300μl超純水封住上表面。
室溫放置30分鐘以上,待水和凝固的分離膠兩相界面變清晰,準備好5%濃縮膠(ddH2O:2.7ml,30%acrylamide mix:0.67ml,Tris(PH 6.8 0.5M):0.5ml,10%SDS:40μl,10%APS:40μl,TEMED:4μl),倒掉玻璃板中的水,注入濃縮膠,插上梳子,室溫放置20分鐘待其凝固後即可使用。
電泳:
配製10×電泳液(Tris:30.3g,甘氨酸:144.0g,SDS:10.0g,去離子水: 定容至1000ml),將50ml 10×電泳液與450ml H2O2混合即得到500ml 1×電泳液。
安裝好電泳裝置並用清水衝洗乾淨,拔掉梳子。倒上500ml電泳液,保持電泳槽內槽充滿電泳液,樣品在6000轉/分條件下離心1s收集後加入泳道,鄰近樣品放的其中一個空白孔加入預染marker(加拿大Fermentas公司生產,貨號SM0671)。電泳時電源限定條件:恆流15-20mA。待指示劑顯示樣品接近凝膠下邊界時停止電泳。
轉膜:
配製10×轉膜液(Tris:30.3g,甘氨酸:144.0g,H2O:定容至:1000ml)。
接下來配製好1×轉膜液(10×轉膜液:80ml,甲醇:160ml,H2O:定容至800ml)。準備好轉膜裝置,兩組9×8cm規格濾紙,每組三張。把PVDF膜先浸泡在甲醇中約20秒待其浸透無白點,再放入轉膜液中平衡後,轉移至裝有轉膜液的小盒中備用。然後用塑料片輕輕切去濃縮膠,再切割分離膠兩端和與玻璃接觸的面。用塑料片蘸少許轉膜液,塞入分離膠下使其鬆動,然後將其倒扣至裝有轉膜液的塑料盆中輕輕搖晃,分離膠即從玻璃上脫離到轉膜液中。
打開轉膜用的塑料板,浸沒到轉膜液中,在轉膜液中按如下順序組裝:負極板(黑)放上海綿→一組濾紙(三張)→分離膠→PVDF膜→一組濾紙(三張)→海綿→正極板。然後夾持好裝置,倒上轉膜液,確保轉膜的三明治樣結構完全浸沒在轉膜液中,蓋上帶有電源線的蓋子,整個電泳裝置置於塑料盆中,電泳盒外圍加上冰水混合物即可開始轉膜。轉膜時電源限定條件:U=100V;I=350mA;t=75min。
封閉:
配製好10×TBS緩衝液(1L含Tris 24.23g、NaCl 80.06g,HCl調pH至7.6),然後用去離子水稀釋10倍得到1×TBS,再加入0.5%(v/v)的吐溫- 20配製成TBST。準備好5%脫脂奶,將5g脫脂牛奶(中國內蒙古伊利實業集團股份有限公司)溶於100ml TBST溶液中。轉膜完畢後,取出PVDF膜裝入自封袋,倒入20ml的5%脫脂牛奶,用封口機封牢,置於脫色搖床上搖20分鐘,再轉至4℃冰箱過夜存放。
一抗孵育:
從冰箱取出PVDF膜後,先用1×TBST洗滌3次,每次5分鐘。按1:1000稀釋率取用抗體TSPAN8(英國Abcam公司生產,ab22453)及1:8000稀釋率取用抗體HRP偶聯的GAPDH(上海康成公司生產,KC-5G5),加入到5ml TBST中,搖勻即得到抗體工作液。把洗滌後的膜裝入自封袋,倒入配好的抗體工作液,中速運轉的脫色搖床上孵育120分鐘。一抗孵育完後,若一抗上已經有辣根過氧化物酶標記(如內參蛋白GAPDH的抗體),則直接跳至顯影步驟,若沒有,則需要二抗孵育。
二抗孵育:
先用1×TBST洗滌3次,每次5分鐘。吸取辣根過氧化物酶標記的兔免疫球蛋白抗體(即二抗,美國Cell Signaling Technology公司生產,稀釋率為1:2000),加入到5ml 1×TBST中搖勻。把洗滌後的膜裝入自封袋,倒入配好的二抗工作液,封口機封住,搖床上搖120分鐘。
顯影:
二抗孵育結束後,用1×TBST洗滌6次,前3次每次10分鐘,後3次每次5分鐘。在此期間準備好顯影液、定影液、自來水、膠片、顯影暗匣,剪刀,衛生紙。以下操作在顯影暗室中紅光燈輔助下完成。分別吸取400μl顯影底物A和B(中國天根生化科技有限公司生產)混勻,平衡至室溫。把PVDF膜放在潔淨的剪開的自封袋內表面上。迅速加上顯影底物,孵育1-5分鐘,若肉眼可見明顯條帶,則終止孵育。棄掉顯影底物液體,把膜轉移至三面剪開的自封袋中,合上自封袋,用衛生紙小心吸掉多餘的液體,放入顯影暗匣,用絕緣膠帶固定。放上膠片,膠片曝光一定時間(5~30秒)後,依 次放入顯影液、定影液各20秒,然後把膠片放在清水中。最後在自來水下洗淨膠片,晾乾,掃描至計算機,得到檢測結果。
顯影后的膜用1×TBST洗10分鐘後,可放入有抗體洗脫緩衝液的自封袋中,密封后放入50~60℃的水浴鍋中孵育30分鐘。再用1×TBST洗2次,每次10分鐘。洗完後用脫脂奶封閉即可重複上面的步驟檢測其它蛋白如內參蛋白GAPDH。
Western Blot的結果如圖1所示,表明本發明用到的幹擾RNA(Y2038)能顯著降低TSPAN8的蛋白水平。
經實驗發現:siRNA2的幹擾效果最好。shRNA2(Y 2038)的幹擾效果最好。
上述的siRNA序列的shRNA序列,包括正義鏈和反義鏈,此shRNA序列為:
正義鏈:5』-CCGGGCAAUGACUCUCAAGCAACUCGAGUUGCUUGAGAGUCAUUGC-3』(SEQ ID NO.3)
反義鏈:5』-
AAUUCAAAAAAGCAAUGACUCUCAAGCAACUCGAGUUGCUUGAGAGUCAUUGC-3』(SEQ ID NO.4)
實施例4:MTT法檢測幹擾TSPAN8後對U87和U251細胞系增殖的影響
膠質瘤U87細胞系、膠質瘤U251細胞系,購自中科院細胞所。
在直徑6cm培養皿中培養細胞至大約30%匯合度,分別加入對照慢病毒感染顆粒(標記為Negative control組)和幹擾TSPAN8的病毒感染顆粒(標記為Y2038組,感染複數為10),以未加入病毒的細胞培養為空白對照組(標記為Blank組),繼續培養細胞4天。後用96孔板(美國Corning公司生產)培養,每孔約1500個細胞,每孔培養基150μl。分別在如圖3所示的 時間點,每孔加入15μl濃度為5mg/ml的MTT(上海生工生物工程公司生產)繼續孵育4小時。然後吸掉培養物,每孔加入150μl DMSO,在酶標儀(美國Bio-Rad公司生產,iMark168-1130型)的490nm波長下測定吸光度。
結果如圖2所示,表明幹擾TSPAN8能明顯抑制U87、U251細胞的生長。
實施例5:Transwell法檢測幹擾TSPAN8後對U87和U251細胞系侵襲的影響
Transwell侵襲實驗使用Matrigel Invasion Chamber(8μm,24-well cell culture inserts)(BD Biosience公司)。預先用無菌鑷子將侵襲小室夾持懸掛在24孔培養板上,室溫下放置30分鐘,使基質膠凝固。在小室上方加入500μl無血清培養基,37℃孵育2小時,以水化基質膠。消化細胞,通過細胞計數,用無血清培養基配置1x105個細胞/ml的細胞懸液。吸去侵襲小室上方的無血清培養基,加入500μl血清培養基。培養22個小時後,用棉籤擦盡小室中膜正面的細胞,用甲醇固定膜背面的細胞15分鐘,再用0.5%結晶紫染色15分鐘。對於U87和U251細胞,在100x鏡下隨機拍攝9個視野;對於C6細胞,在200x鏡下隨機拍攝9個視野。得到9個視野中細胞數的平均值。
遷移實驗結果如圖3所示,表明幹擾TSPAN8能明顯抑制U87和U251細胞侵襲結果。
實施例6:Transwell法檢測幹擾TSPAN8後對U87和U251細胞系遷移的影響
Transwell侵襲遷移實驗使用孔徑8μm的24孔細胞培養池(BD Biosience公司)。與侵襲實驗的區別在於無基質膠包被,無凝固和水化步驟。用無血清培養基配置5x105個細胞/ml的細胞懸液,在小室上方加入100μl懸液,即5x104個細胞,並在小室下方加入500μl血清培養基。培養4小時後結束實 驗。後續處理同Transwell侵襲實驗。實驗結果如圖4所示,表明幹擾TSPAN8能明顯抑制U87和U251細胞的遷移。
上述實施例為本發明較佳的實施方式,但本發明的實施方式不受上述實施例的限制。其他任何不脫離本發明之精神和原理下所作的變形,均應認為是本發明的保護範圍。