氨甲環酸環烯烴雜質的合成方法及氨甲環酸環烯烴雜質的應用與流程
2023-10-11 17:27:24 3
本發明涉及化學合成
技術領域:
,尤其是涉及一種氨甲環酸環烯烴雜質的合成方法及氨甲環酸環烯烴雜質的應用。
背景技術:
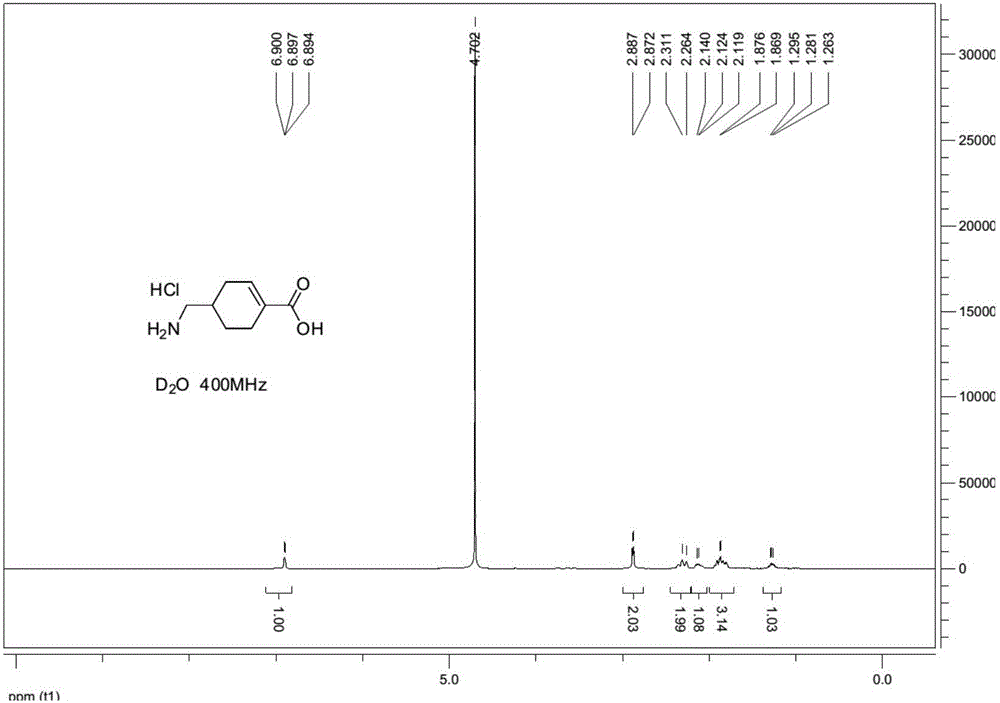
:氨甲環酸,又稱傳明酸,其英文名稱為:TranexamicAcid,其化學名稱為:對氨甲基環己烷甲酸或者反式-4-氨甲基-環己烷甲酸。氨甲環酸主要用於主要用於急性或慢性、局限性或全身性纖維蛋白溶解亢進所致的各種出血。氨甲環酸的質量標準在我國藥典中有所記載,其中明確指出可能含有環烯烴雜質,英國藥典也明確記載了氨甲環酸中環烯烴雜質的結構。現有技術中,已經公開了多種氨甲環酸質量控制的檢測方法,以檢測包括氨甲環酸環烯烴在內的多種雜質的含量,但是現有技術中,尚未公開氨甲環酸環烯烴雜質的高效合成方法。氨甲環酸環烯烴雜質是氨甲環酸質量控制中非常重要的雜質,對氨甲環酸雜質的相關研究意義重大。因此,提供一種氨甲環酸環烯烴雜質的高效合成方法,能夠彌補現有技術中的不足,為氨甲環酸的質量控制提供合格、廉潔、易得的對照品,為安全用藥提供重要的指導意義。因此,特提出本發明。技術實現要素:本發明的目的在於提供氨甲環酸環烯烴雜質的合成方法,從而為氨甲環酸的質量控制提供雜質對照品。本發明提供了一種氨甲環酸環烯烴雜質的合成方法,所述合成反應包括以下步驟:(1)式I所示結構的化合物和鄰苯二甲酸酐,在第一有機溶劑中發生醯胺化反應,生成式II所示結構的化合物;(2)式II所示結構的化合物和二氯亞碸,在第二有機溶劑中發生醯氯化反應,得到式III所示結構的化合物;(3)式III所示結構的化合物和溴,在第三有機溶劑中發生取代反應,得到式IV所示結構的化合物;(4)式IV所示結構的化合物和有機醇,發生酯化反應,得到式V所示結構的化合物;(5)式V所示結構的化合物,在脫滷化氫試劑作用下,在第五有機溶劑中發生脫滷化氫反應,得到式VI所示結構的化合物;(6)式VI所示結構的化合物和濃鹽酸,發生水解反應,得到式VII所示結構的化合物,即氨甲環酸環烯烴雜質,所述氨甲環酸環烯烴雜質為式VII所示結構的化合物;其中,R基團為C1-C5的烷基。進一步的,步驟(2)中,所述醯氯化反應中還加入催化劑N,N-二甲基甲醯胺;步驟(3)中,所述取代反應中還加入催化劑紅磷。進一步的,步驟(1)中,所述第一有機溶劑為冰乙酸;步驟(2)中,所述第二有機溶劑為二氯乙烷或者氯仿;步驟(3)中,所述第三有機溶劑為二氯乙烷或者氯仿;步驟(4)中,所述有機醇為甲醇或者乙醇,所述R基團為甲基或者乙基;步驟(5)中,所述第五有機溶劑為四氫呋喃或者1,4-二氧六環,所述脫滷化氫試劑為1,8-二氮雜雙環[5.4.0]十一碳-7-烯;步驟(6)中,所述水解反應在第六有機溶劑中進行,所述第六有機溶劑為1,4-二氧六環。進一步的,步驟(1)中,所述醯胺化反應的反應溫度為115-120℃;步驟(2)中,所述醯氯化反應的反應溫度為80-85℃;步驟(3)中,所述取代反應的反應溫度為80-85℃;步驟(4)中,所述酯化反應的反應溫度為45-55℃;步驟(5)中,所述脫滷化氫反應的反應溫度為65-70℃;步驟(6)中,所述水解反應的反應溫度為110-120℃。進一步的,步驟(1)中,所述醯胺化反應的反應溫度為118℃;步驟(2)中,所述醯氯化反應的反應溫度為83℃;步驟(3)中,所述取代反應的反應溫度為83℃;步驟(4)中,所述酯化反應的反應溫度為50℃;步驟(5)中,所述脫滷化氫反應的反應溫度為67℃;步驟(6)中,所述水解反應的反應溫度為115℃。進一步的,步驟(1)中,所述式I所示結構的化合物、所述鄰苯二甲酸酐、所述第一有機溶劑的摩爾數比為1:0.8-1.2:20-40;步驟(2)中,所述式II所示結構的化合物、所述二氯亞碸、所述第二有機溶劑、所述N,N-二甲基甲醯胺的摩爾數比為1:2-3:8-15:0.02-0.06;步驟(3)中,所述式III所示結構的化合物、所述溴、所述紅磷的摩爾數比為1:2-2.5:0.3-0.8;步驟(4)中,所述式IV所示結構的化合物、所述有機醇的摩爾數比為1:90-100;步驟(5)中,所述式V所示結構的化合物、所述脫滷化氫試劑、所述第五有機溶劑的摩爾數比為1:1.3-2.0:50-70;步驟(6)中,所述式VI所示結構的化合物、所述濃鹽酸、所述第六有機溶劑的摩爾數比為1:8-12:38-45。進一步的,步驟(1)中,所述式I所示結構的化合物、所述鄰苯二甲酸酐、所述冰乙酸的質量為1:1:27;步驟(2)中,所述式II所示結構的化合物、所述二氯亞碸、所述二氯乙烷、所述N,N-二甲基甲醯胺的質量比為1:2.4:11:0.04;步驟(3)中,所述式III所示結構的化合物、所述溴、所述紅磷的質量比為1:2.2:0.5;步驟(4)中,所述式IV所示結構的化合物、所述甲醇的質量比為1:95;步驟(5)中,所述式V所示結構的化合物、所述脫滷化氫試劑、所述四氫呋喃的質量比為1:1.75:56;步驟(6)中,所述式VI所示結構的化合物、所述濃鹽酸、所述四氫呋喃的質量比為1:9.7:42.4。進一步的,步驟(1)中,所述醯胺化反應的反應時間為2.5-3.5h;步驟(2)中,所述醯氯化反應的反應時間為1-2h;步驟(3)中,所述取代反應的反應時間為100-150h;步驟(4)中,所述酯化反應的反應時間為0.5-1.5h;步驟(5)中,所述脫滷化氫反應的反應時間為50-70h;步驟(6)中,所述水解反應的反應時間為100-200h。進一步的,步驟(1)中,所述醯胺化反應的反應時間為3h;步驟(2)中,所述醯氯化反應的反應時間為1.5h;步驟(3)中,所述取代反應的反應時間為130h;步驟(4)中,所述酯化反應的反應時間為1h;步驟(5)中,所述脫滷化氫反應的反應時間為60h;步驟(6)中,所述水解反應的反應時間為120h。另外本發明的另一目的在於提供一種按照所述的合成方法合成的氨甲環酸環烯烴雜質在製備氨甲環酸雜質對照品中的應用,以為氨甲環酸的質量控制提供雜質對照品。本發明提供的氨甲環酸環烯烴雜質的合成方法以氨甲環酸和鄰苯二甲酸酐為原料,經過醯胺化反應、醯氯化反應、取代反應、酯化反應、脫滷化氫反應、水解反應等步驟,能夠得到純度高、結構正確的氨甲環酸環烯烴雜質。另外,本發明提供了按照所述方法合成的氨甲環酸雜質在製備氨甲環酸雜質對照品中的應用,為氨甲環酸的質量控制提供了合格、廉價、易得的雜質對照品。附圖說明為了更清楚地說明本發明具體實施方式或現有技術中的技術方案,下面將對具體實施方式或現有技術描述中所需要使用的附圖作簡單地介紹,顯而易見地,下面描述中的附圖是本發明的一些實施方式,對於本領域普通技術人員來講,在不付出創造性勞動的前提下,還可以根據這些附圖獲得其他的附圖。圖1為本發明實施例6合成的化合物VII的氫譜圖。具體實施方式下面將結合附圖對本發明的技術方案進行清楚、完整地描述,顯然,所描述的實施例是本發明一部分實施例,而不是全部的實施例。基於本發明中的實施例,本領域普通技術人員在沒有做出創造性勞動前提下所獲得的所有其他實施例,都屬於本發明保護的範圍。本發明提供了一種氨甲環酸環烯烴雜質的合成方法,包括以下步驟:(1)式I所示結構的化合物和鄰苯二甲酸酐,在第一有機溶劑中發生醯胺化反應,生成式II所示結構的化合物;(2)式II所示結構的化合物和二氯亞碸,在第二有機溶劑中發生醯氯化反應,得到式III所示結構的化合物;(3)式III所示結構的化合物和溴,在第三有機溶劑中發生取代反應,得到式IV所示結構的化合物;(4)式IV所示結構的化合物和有機醇,發生酯化反應,得到式V所示結構的化合物;(5)式V所示結構的化合物,在脫滷化氫試劑作用下,在第五有機溶劑中發生脫滷化氫反應,得到式VI所示結構的化合物;(6)式VI所示結構的化合物和濃鹽酸,發生水解反應,得到式VII所示結構的化合物,即氨甲環酸環烯烴雜質。在一個優選的實施方式中,步驟(1)還包括對反應生成的式II所示結構的化合物的純化,具體為:醯胺化反應結束後,待反應液降至室溫,加水,析出固體過濾、烘乾,得到的白色固體即為式II所示結構的化合物。其中加入水的體積(mL)為式I所示結構的化合物的質量(g)的10-20倍,優選15倍。在另一個優選的實施方式中,步驟(4)還包括對反應生成的式V所示結構的化合物的純化,具體為:酯化反應結束後,反應液減壓蒸餾除去甲醇,向反應液中加入乙酸乙酯,依次用水、飽和碳酸氫鈉溶液、飽和氯化鈉溶液洗滌,再用無水硫酸鈉乾燥、過濾,濃縮幹,得到的淡黃色固體即為式V所示結構的化合物。在另一個優選的實施方式中,步驟(5)還包括對反應生成的式VI所示結構的化合物的純化,具體為:託滷化氫反應結束後,待反應液降至室溫,向溶液中加入同體積的石油醚,墊矽膠過濾,再用二氯甲烷和石油醚的混合溶劑衝洗矽膠柱,將所有溶液濃縮幹,得到的白色固體即為式VI所示結構的化合物。在另一個優選的實施方式中,步驟(6)還包括對反應生成的式VII所示結構的化合物的純化,具體為:水解反應結束後,將反應液濃縮除去1,4-二氧六環,加少量水,用乙酸乙酯萃取,水層濃縮幹,即得到式VII所示結構的化合物。在另一個優選實施方式中,步驟(4)中的有機醇為甲醇,R基團為甲基,式IV所示結構的化合物和甲醇,發生酯化反應,得到式V-1所示結構的化合物。式V-1所示結構的化合物經過脫滷化氫反應,生成式VI-1所示結構的化合物。在另一個優選實施方式中,步驟(4)中的有機醇為乙醇,R基團為乙基,式IV所示結構的化合物和甲醇,發生酯化反應,得到式V-2所示結構的化合物。式V-2所示結構的化合物經過脫滷化氫反應,生成式VI-2所示結構的化合物。為了有助於更清楚的理解本發明,現結合具體實施例解釋如下。如未明確指出,實施例中用到的原料試劑均由市場購得,實施例中出現的一些試劑的中英文對照如表1所示。表1試劑中英文對照中文全稱英文縮寫N,N-二甲基甲醯胺DMF冰乙酸AcOH二氯亞碸SOCl2二氯乙烷DCE甲醇MeOH1,8-二氮雜雙環[5.4.0]十一碳-7-烯DBU四氫呋喃THF1,4-二氧六環DOA乙醇EtOH實施例1化合物II的製備在1000mL燒瓶內,加入40g化合物I,37.7g鄰苯二甲酸酐,400mL冰乙酸。118℃回流反應3小時,TLC點板顯示原料反應完全,其中TLC展開劑為冰乙酸:甲醇=1:4。待反應液降至室溫,向反應液中加入600mL冰水,反應液中有固體析出,將析出的固體過濾、水洗、烘乾,得到白色固體即為化合物II,其質量為70g,收率為95.8%。實施例2化合物III的製備在1000mL燒瓶內,加入40g化合物II,400mL二氯乙烷,3滴DMF,攪拌下,緩慢加入40g二氯亞碸。二氯亞碸加完後,83℃回流反應1.5小時,TLC點板顯示化合物II反應完全,其中TLC展開劑為石油醚:乙酸乙酯=5:1。其中,溶劑二氯乙烷也可以用氯仿替代。實施例3化合物IV的製備實施例2的反應完成後,反應液降溫至60℃以下,小心緩慢加入催化量的紅磷,加完,再加入46g溴素,升溫至83℃反應約130h。採用HPLC監控反應,待原料基本反應完時,反應結束。實施例4化合物V-1的製備實施例3的反應完成後,反應液降溫至50℃,緩慢加入400mL甲醇,50℃反應1h。TLC點板顯示原料反應完全,其中TLC展開劑為石油醚:乙酸乙酯=5:1。濃縮除去大部分的甲醇,用乙酸乙酯溶解,水洗,飽和碳酸氫鈉溶液洗,飽和食鹽水洗,無水硫酸鈉乾燥,過濾,濃縮幹,得到的淡黃色固體即為化合物V-1,其質量為50g,收率為94.45%。實施例5化合物VI-1的製備在1000ml燒瓶內,加入50g化合物V-1,600mL四氫呋喃和35gDBU,67℃回流反應60h。HPLC顯示原料反應完。反應液降至室溫過濾,然後用二氯甲烷:石油醚=1:1的溶液過快速柱,濃縮幹,得到的白色固體即為化合物VI-1,其質量為28g,收率為71.1%。其中,溶劑四氫呋喃也可以用1,4-二氧六環替代。實施例6化合物VII的製備在250ml燒瓶內,加入5g化合物VI-1、60mL1,4-二氧六環和30mL濃鹽酸,115℃回流反應120h,中間補加濃鹽酸20mL。TLC點板顯示化合物VII反應完全,其中TLC展開劑為石油醚:乙酸乙酯=5:1。反應液濃縮出去大部分1,4-二氧六環,加少量水,用乙酸乙酯萃取5次,水層濃縮幹,得到固體即為化合物VII粗品,共3.3g。用甲叔基丁基醚:乙醇=3:1打漿,過濾得到類白色樣品即為化合物VII,其質量為2.5g,收率為78%。圖1為按照該方法合成的化合物VII的氫譜圖,表明化合物VII即為氨甲環酸環烯烴雜質。實施例7化合物V-2的製備實施例3的反應完成後,反應液降溫至50℃,緩慢加入400mL乙醇,50℃反應1h。TLC點板顯示原料反應完全,其中TLC展開劑為石油醚:乙酸乙酯=5:1。濃縮除去大部分的乙醇,用乙酸乙酯溶解,水洗,飽和碳酸氫鈉溶液洗,飽和食鹽水洗,無水硫酸鈉乾燥,過濾,濃縮幹,得到的淡黃色固體即為化合物V-2,其質量為52g,收率為95%。實施例8化合物VI-2的製備在1000ml燒瓶內,加入52g化合物V-2,600mL四氫呋喃和35gDBU,67℃回流反應60h。HPLC顯示原料反應完。反應液降至室溫過濾,然後用二氯甲烷:石油醚=1:1的溶液過快速柱,濃縮幹,得到的白色固體即為化合物VI-2,其質量為29g,收率為70%。其中,溶劑四氫呋喃也可以用1,4-二氧六環替代。實施例9化合物VII的製備在250ml燒瓶內,加入5.2g化合物VI-2、60mL1,4-二氧六環和30mL濃鹽酸,115℃回流反應約120h,中間補加濃鹽酸20mL。TLC點板顯示化合物VII反應完全,其中TLC展開劑為石油醚:乙酸乙酯=5:1。反應液濃縮出去大部分1,4-二氧六環,加少量水,用乙酸乙酯萃取5次,水層濃縮幹,得到固體即為化合物VII粗品,共3.2g。用甲叔基丁基醚:乙醇=3:1打漿,過濾得到類白色樣品即為化合物VII,其質量為2.4g,收率為77%。最後應說明的是:以上各實施例僅用以說明本發明的技術方案,而非對其限制;儘管參照前述各實施例對本發明進行了詳細的說明,本領域的普通技術人員應當理解:其依然可以對前述各實施例所記載的技術方案進行修改,或者對其中部分或者全部技術特徵進行等同替換;而這些修改或者替換,並不使相應技術方案的本質脫離本發明各實施例技術方案的範圍。當前第1頁1 2 3