螺雙芴衍生物及其在有機電致發光領域中的應用的製作方法
2023-09-27 07:55:55 3
本發明屬於有機光電材料技術領域,涉及間位取代螺雙芴化合物和本發明化合物在電子器件特別是在有機電致發光器件、有機場效應電晶體、有機太陽能電池中的用途,尤其是在有機電致發光器件中用作發光主體材料、激子阻擋材料或電子傳輸材料,另外還涉及包含所述化合物的電子器件。
背景技術:
滿足各種信息顯示需求的發光材料在資訊時代的今天發展尤其迅速。發光材料主要分為無機和有機
兩類,與無機材料相比,有機材料具有相對更高的發光效率和更寬的發光顏色選擇範圍以及具有大面積成膜的優勢,引起研究者極大的興趣。1987年柯達公司c.w.tang等人首次報導以真空蒸鍍的方法製備出alq3為發光材料的雙層發光器件,有力地推動了有機電致發光材料的發展。經過30多年的發展,有機電致發光在平板顯示、照明方面的應用已經進入產業化初級階段。從發光材料來分類,有機發光二極體(organiclight-emittingdiode,oled)可以分為螢光、磷光以及熱激發延遲螢光發光二極體。
有機電致發光器件一般包括由陽極、金屬陰極和它們之間夾著的有機層組成。有機層主要有空穴注入層、空穴傳輸層、電子阻擋層、發光層、空穴阻擋層、電子傳輸層、電子注入層。另外,發光層大多採用主客體結構,即將發光材料以一定濃度摻雜在主體材料中,以避免濃度淬滅和三線態-三線態湮滅,提高發光效率。因此一般要求主體材料具有較高的三線態能級,並且同時具有較高的穩定性。
現有技術中主體材料多採用咔唑基團,可使得材料具有高額空穴傳輸能力,但咔唑基團c-n鍵容易分解,影響器件的耐久性。為了改善器件的穩定性,有報導設計具有螺環連接鍵的材料可具有加強的形態穩定性。螺雙芴分子具有非平面的空間結構,兩個芴單體以sp3雜化的c原子為中心橋聯在一起,將其引入到具有電致發光特性的分子中,對於提高分子的熱穩定性和光譜穩定性等方面具有很大的應用價值。目前,對於螺雙芴衍生物的研究主要集中在其2位和4位(如cn102077384a和cn104541576a)。然而,2位和4位的衍生物一般具有較大的共軛度,分子的三重態能級較低,大大限制了其在藍色有機電致發光器件中的應用。
技術實現要素:
發明要解決的問題
為了解決以上現有技術的缺點和不足之處,有效提高三線態能級,改善有機電致發光器件的壽命、效率和工作電壓等性能,本發明的目的是提供一種間位取代螺雙芴化合物以及製備方法。
本發明的另一目的在於提供上述螺雙芴類化合物的一種應用,特別是在有機電致發光器件中的應用。
本發明的再一目的在於提供一種包含所述化合物的電子器件。
用於解決問題的方案
令人驚訝地發現,下面描述的化合物能有效提高三線態能級,能夠有效改進有機電致發光器件的壽命、效率和工作電壓等性能。
本發明涉及式(ⅰ)或式(ⅱ)的化合物
其中,
螺雙芴母核通過l1、l2分別連接1個或2個r1或r2基團,-l1-r1和/或-l2-r2以碳原子連接到螺雙芴母核3位和/或6位;
l1、l2分別獨立地選自單鍵、羰基、具有6至18個環原子的芳族或雜芳族環系;
r1、r2分別獨立地為具有6至30個環原子的芳族或雜芳族環系,所述芳族或雜芳族環系可任選地被一個或多個基團r1取代;
r1:在每次出現時相同或不同地選自:氫、氘、滷素、氰基;具有1至40個c原子的直鏈烷基、烷氧基或硫代烷基基團或者具有3至40個c原子的支鏈或環狀的烷基、烷氧基或硫代烷基基團,所述直鏈烷基或支鏈烷基可任選地被滷素取代;具有6至30個環原子的芳族環系或具有5至30個環原子的雜芳族環系;
當化合物為式(ⅰ)時,若l1為單鍵或羰基時,螺雙芴母核通過l1連接1個r1;若l1為具有6至18個環原子的芳族或雜芳族環系時,螺雙芴母核通過l1連接2個r1,每個r1可相同或不同;
當化合物為式(ⅱ)時,若l1、l2分別獨立地為單鍵或羰基時,螺雙芴母核通過l1連接1個r1和r2;若l1、l2分別獨立地為具有6至18個環原子的芳族或雜芳族環系時,螺雙芴母核通過l1、l2分別連接2個r1和r2,每個r1、每個r2可相同或不同;
所述化合物不包括式a所示結構的化合物:
優選地,l1、l2分別獨立地為單鍵、羰基、苯基、三嗪基或聯苯基;雜芳族環系中雜原子選自n、o和/或s。
優選地,r1、r2分別獨立地選自如下結構(ar-1至ar-29):
其中虛線表示與l1或l2鍵合的鍵,r1具有上述含義。
優選地,l1、l2分別獨立地為單鍵、羰基、苯基或三嗪基;
r1、r2分別獨立地選自以下芳族或雜芳族環系:苯基、聯苯基、三聯苯基、四聯苯基、五聯苯基、苯並蒽、苯並菲、芴基、螺雙芴基、三嗪基、二苯並呋喃基、二苯並噻吩基、咔唑基、n-苯基咔唑基、茚並咔唑基、苯並咪唑基、二苯基-苯並咪唑基、二苯基-噁二唑基,所述芳族或雜芳族環系可被一個或多個r1取代;
r1在每次出現時相同或不同地選自:氫、具有1至5個c原子的直鏈烷基、苯基、苯並菲、聯苯基、三聯苯基、四聯苯基、五聯苯基、芴基、喹啉基、螺雙芴基、三嗪基、二苯並呋喃基、二苯並噻吩基、咔唑基、茚並咔唑基、苯並咪唑基。
進一步優選地,l1、l2分別獨立地為單鍵、羰基或三嗪基;
r1、r2分別獨立地選自以下芳族或雜芳族環系:苯基、苯並菲、螺雙芴基、三嗪基;所述芳族或雜芳族環系可被一個或多個r1取代,每個r1、每個r2不同時為苯基;r1在每次出現時相同或不同地選自:氫、具有1至5個c原子的直鏈烷基、苯基、苯並菲、聯苯基、三聯苯基、四聯苯基、五聯苯基、芴基、螺雙芴基、三嗪基、二苯並呋喃基、二苯並噻吩基、咔唑基、苯並咪唑基。
進一步優選地,本發明化合物選自:
本發明還提供一種用於製備以上任一項化合物的方法,其特徵在於通過3位和/或6位官能化的螺雙芴和含有官能化基團-l1-r1和/或-l2-r2的化合物進行金屬催化偶聯反應引入基團-l1-r1和/或-l2-r2。
更進一步地,本發明提供上述化合物在電子器件特別是在有機電致發光器件、有機場效應電晶體、有機太陽能電池中的用途,尤其是製備有機電致發光器件中的發光主體材料、激子阻擋材料或電子傳輸材料的用途。
本發明還提供一種電子器件,其包括至少一種上述化合物。優選地,該電子器件是有機電致發光器件,所述有機電致發光器件包含有機發光層、激子阻擋層、電子傳輸層,其中任意一層或多層含有上述任一種化合物。
發明的效果
本發明的3位和/或6位取代的螺雙芴衍生物的三線態能級高達2.80ev,較2位(2.54ev)和4位(2.65ev)取代的螺雙芴衍生物的三線態高。因此其可作為主體材料、激子阻擋材料或者電子傳輸材料應用在電致發光器件中,其能有效抑制能量的耗損,有望實現高效率、長壽命的器件性能。本發明以環金屬銥配合物ir(ppy)3為客體製備的發光器件,在10000坎特拉每平方米下,最大外量子效率可達16.8%,同時在10000坎特拉每平方米下,器件保持了極高的穩定性,在衰減到起始亮度的90%時,壽命長達1000個小時。除此之外,以熱激發延遲螢光材料4czipn為客體製備的發光器件,在10000坎特拉每平方米下,最大外量子效率可達15.4%,同時在10000坎特拉每平方米下,在衰減到起始亮度的90%時,壽命長達800個小時。另外,以藍色螢光材料bd1為客體製備的發光器件,在5000坎特拉每平方米下,最大外量子效率可達5.2%,同時在5000坎特拉每平方米下,在衰減到起始亮度的90%時,壽命長達600個小時。而已報導的2位取代的螺雙芴衍生物為主體材料並以藍色螢光材料bd1為客體製備的發光器件,其最大外量子效率僅為2.0%,同時在5000坎特拉每平方米下,在衰減到起始亮度的90%時,壽命為100個小時。由此可見本發明具有有效的技術效果。
附圖說明
圖1為化合物1-75的紫外吸收光譜(uv-vis)、室溫螢光光譜(pl)以及低溫磷光光譜(phos)。
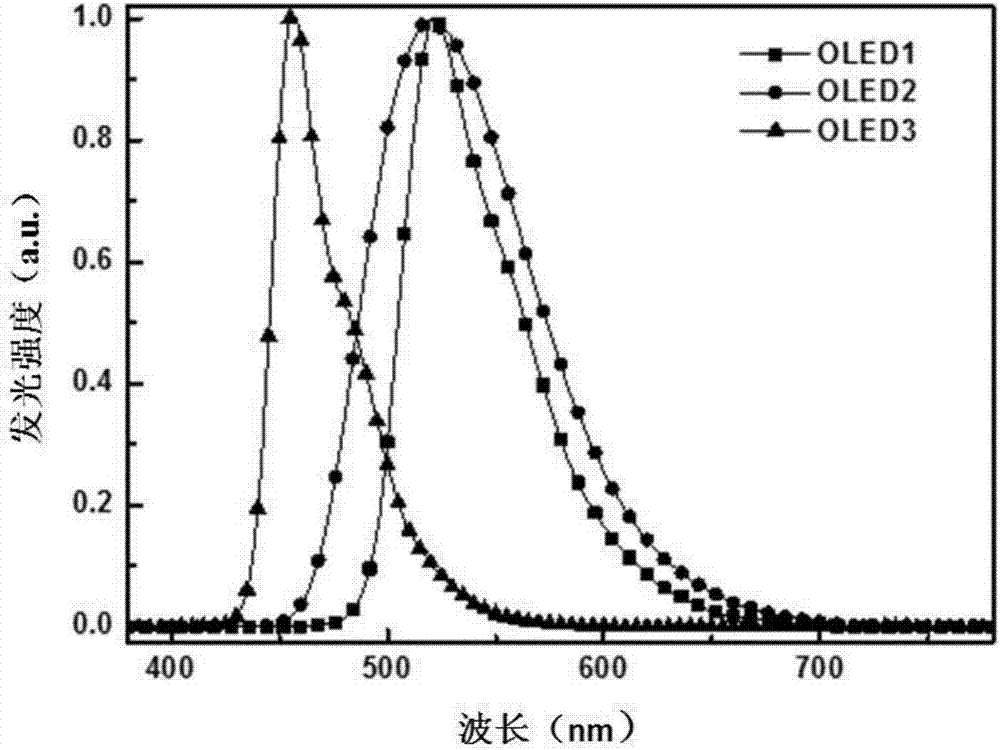
圖2為實施例oled1、oled2和oled3的有機電致發光光譜。
具體實施方式
下面將結合實施例對本發明的技術方案進行詳細的描述。
出於本發明的目的,本發明意義上的芳族環系在該環系中含有6至30個c原子,優選6至18個c原子。本發明意義上的雜芳族環系在該環系中含有5至30個,優選6至18個c原子和雜原子,該雜原子優選地選自n、o和/或s。在本發明中,化合物的雜原子數目可以是1-5個(取整數)。本發明意義上的芳族或雜芳族環系旨在被認為是指如下的體系,該體系並不必須僅含有芳基或雜芳基基團,而是其中多個芳基或雜芳基基團還可以被非芳族單元(優選小於非氫原子的10%)間斷,該非芳族單元例如是c、n或o原子或者羰基基團或者-c(cf3)2-。例如,和兩個或更多個芳基基團例如被直鏈或環狀的烷基基團或者被甲矽烷基基團間斷的體系一樣,9,9』-螺二芴、9,9-二芳基芴、三芳基胺、二芳基醚等的體系也旨在被認為是在本發明意義上的芳族環系。此外,其中兩個或更多個芳基或雜芳基基團相互直接鍵合的體系,例如聯苯、三聯苯或四聯苯,同樣旨在被認為是芳族或雜芳族環系。
芳族或雜芳族環系例如由衍生自以下基團的一種或多種所構成的體系:苯、萘、蒽、苯並蒽、菲、苯並菲、芘、苝、熒蒽、苯並熒蒽、並四苯、並五苯、苯並芘、聯苯、偶苯、三聯苯、四聯苯、五聯苯、三聚苯、芴、螺雙芴、二氫菲、二氫芘、四氫芘、順式或反式茚並芴、順式或反式單苯並茚並芴、順式或反式二苯並茚並芴、三聚茚、異三聚茚、螺三聚茚、螺異三聚茚、呋喃、苯並呋喃、異苯並呋喃、二苯並呋喃、噻吩、苯並噻吩、異苯並噻吩、二苯並噻吩、吡咯、吲哚、異吲哚、咔唑、吲哚並咔唑、茚並咔唑、吡啶、喹啉、異喹啉、吖啶、菲啶、苯並-5,6-喹啉、苯並-6,7-喹啉、苯並-7,8-喹啉、吩噻嗪、吩噁嗪、吡唑、吲唑、咪唑、苯並咪唑、萘並咪唑、菲並咪唑、吡啶並咪唑、吡嗪並咪唑、喹喔啉並咪唑、噁唑、苯並噁唑、萘並噁唑、蒽並噁唑、菲並噁唑、異噁唑、1,2-噻唑、1,3-噻唑、苯並噻唑、噠嗪、苯並噠嗪、嘧啶、苯並嘧啶、喹喔啉、1,5-二氮雜蒽、2,7-二氮雜芘、2,3-二氮雜芘、1,6-二氮雜芘、1,8-二氮雜芘、4,5-二氮雜芘、4,5,9,10-四氮雜苝、吡嗪、吩嗪、吩噁嗪、吩噻嗪、熒紅環、萘啶、氮雜咔唑、苯並咔啉、菲咯啉、1,2,3-三唑、1,2,4-三唑、苯並三唑、1,2,3-噁二唑、1,2,4-噁二唑、1,2,5-噁二唑、1,3,4-噁二唑、1,2,3-噻二唑、1,2,4-噻二唑、1,2,5-噻二唑、1,3,4-噻二唑、1,3,5-三嗪、1,2,4-三嗪、1,2,3-三嗪、四唑、1,2,4,5-四嗪、1,2,3,4-四嗪、1,2,3,5-四嗪、嘌呤、蝶啶、吲嗪、苯並噻二唑等,上述基團可被取代或不被取代。
本發明意義上環狀的烷基、烷氧基或硫代烷氧基基團被認為是指單環、雙環或多環的基團。
在本發明的一種實施方式中,式(ⅰ)或式(ⅱ)的化合物為:
其中,螺雙芴母核通過l1、l2分別連接1個或2個r1或r2基團,-l1-r1和/或-l2-r2以碳原子連接到螺雙芴母核3位和/或6位。取代基r1、r2通過l1、l2連接在螺雙芴母核3位或者6位,這種連接方式可以打斷整個分子的共軛,從而提高分子的三線態能級。
本發明的實施方案可為:
(1)對於l1、l2:
l1、l2分別獨立地選自單鍵、羰基、具有6至18個環原子的芳族或雜芳族環系,所述芳族或雜芳族環系可任選地被一個或多個基團取代;優選地,l1、l2分別獨立地選自單鍵、羰基、具有6至12個環原子的芳族或雜芳族環系;更優選地,l1、l2分別獨立地為單鍵、羰基、苯基、三嗪基或聯苯基。
(2)對於r1、r2:
r1、r2分別獨立地為具有6至30個環原子的芳族或雜芳族環系,所述芳族或雜芳族環系可任選地被一個或多個基團r1取代;
當化合物為式(ⅰ)時,若l1為單鍵或羰基時,螺雙芴母核通過l1連接1個r1;若l1為具有6至18個環原子的芳族或雜芳族環系時,螺雙芴母核通過l1連接2個r1,每個r1可相同或不同;當化合物為式(ⅱ)時,若l1、l2分別獨立地為單鍵或羰基時,螺雙芴母核通過l1、l2連接1個r1和r2;若l1、l2分別獨立地為具有6至18個環原子的芳族或雜芳族環系時,螺雙芴母核通過l1、l2分別連接2個r1和r2,每個r1、每個r2可相同或不同;
優選地,r1、r2分別獨立地選自以下芳族或雜芳族環系:苯基、聯苯基、三聯苯基、四聯苯基、五聯苯基、苯並蒽、苯並菲、芴基、螺雙芴基、三嗪基、二苯並呋喃基、二苯並噻吩基、咔唑基、n-苯基咔唑基、茚並咔唑基、苯並咪唑基、二苯基-苯並咪唑基、二苯基-噁二唑基,所述芳族或雜芳族環系可被一個或多個r1取代;
同樣,優選地,r1、r2分別獨立地選自如上文所述結構ar-1至ar-29;
更優選地,r1、r2分別獨立地選自以下芳族或雜芳族環系:苯基、苯並菲、螺雙芴基、三嗪基;所述芳族或雜芳族環系可被一個或多個r1取代,每個r1、每個r2不同時為苯基。
(3)對於取代基r1:
r1在每次出現時相同或不同地選自:氫、氘、滷素、氰基;具有1至40個c原子的直鏈烷基、烷氧基或硫代烷基基團或者具有3至40個c原子的支鏈或環狀的烷基、烷氧基或硫代烷基基團,所述直鏈烷基或支鏈烷基可任選地被滷素取代;具有6至30個環原子的芳族環系或具有5至30個環原子的雜芳族環系;
優選地,r1在每次出現時相同或不同地選自:氫、氘、氟、氰基;具有1至10個c原子的直鏈烷基、烷氧基或者具有3至10個c原子的支鏈或環狀的烷基或烷氧基,所述直鏈烷基或支鏈烷基可任選地被氟取代;具有6至30個環原子的芳族或雜芳族環系;
更優選地,r1在每次出現時相同或不同地選自:氫、氟;具有1至5個c原子的直鏈烷基如甲基、乙基、丙基、丁基,具有3至6個c原子的支鏈或環狀的烷基,具有6至30個環原子的芳族或雜芳族環系如苯基、苯並菲、聯苯基、三聯苯基、四聯苯基、五聯苯基、芴基、喹啉基、螺雙芴基、三嗪基、二苯並呋喃基、二苯並噻吩基、咔唑基、茚並咔唑基、苯並咪唑基。
在本發明的一種特別優選的實施方式中:
l1、l2分別獨立地為單鍵、羰基或三嗪基;
r1、r2分別獨立地選自以下芳族或雜芳族環系:苯基、苯並菲、螺雙芴基、三嗪基;所述芳族或雜芳族環系可被一個或多個r1取代,每個r1、每個r2不同時為苯基;
r1在每次出現時相同或不同地選自:氫、具有1至5個c原子的直鏈烷基、苯基、苯並菲、聯苯基、三聯苯基、四聯苯基、五聯苯基、芴基、螺雙芴基、三嗪基、二苯並呋喃基、二苯並噻吩基、咔唑基、苯並咪唑基。
本發明合適化合物的實例如化合物1-1到1-135、2-1到2-6中的一種或多種。
可通過本領域普通技術人員已知的合成步驟,例如溴化、鈴木(suzuki)偶聯、布赫瓦爾德-哈特維希(hartwig-buchwald)偶聯等,來製備根據本發明的化合物。
本發明的化合物合成通常從3位和/或6位官能化的螺雙芴衍生物開始,然後通過金屬催化偶聯反應如鈴木(suzuki)偶聯而引入基團-l1-r1和/或-l2-r2。
在本發明的一種優選實施方式中,所述螺雙芴衍生物是利用硼酸衍生物官能化的化合物,且基團-l1-r1和/或-l2-r2源自滷素官能化的化合物。在本發明的另一種優選的實施方式中,所述螺雙芴衍生物是滷素官能化的化合物,且基團-l1-r1和/或-l2-r2源自硼酸衍生物官能化的化合物。
在本發明的第三種優選實施方式中,所述螺雙芴衍生物是滷素官能化的化合物,在格氏試劑的作用下與氰基化的化合物反應而引入基團-l1-r1和/或-l2-r2。其中,滷素官能化優選溴化、氯化、碘化,特別是溴化。
具體而言,如上所述的第一種優選實施方式,需先製備硼酸官能化的螺雙芴衍生物(3-硼酸-9,9』螺雙芴,即中間體1),優選的製備步驟示例如下:
通過控制反應條件,m1的收率可達到80-90%,m2的收率可達到60-77%,中間體1的收率可到80-90%。得到中間體1後,在體系中加入可引入基團-l1-r1和/或-l2-r2的滷素官能化化合物如2,4-二氯-6-苯基-1,3,5-三嗪或三聚氰胺,以及一定量的鈀類催化劑如四(三苯基磷鈀)、無水碳酸鉀、四氫呋喃和水,在氮氣保護下50-80℃下反應30-35小時,反應完畢。蒸去溶劑,用二氯甲烷和水溶解殘留物,水洗,分出有機層,水層用二氯甲烷萃取,合併有機層,用水洗兩次至中性,蒸去溶劑後,柱層析分離,乾燥得到產物。中間體1與四(三苯基磷鈀)的摩爾比在15-20:1,優選18:1;通過調節反應條件,收率在65-85%。
如上所述的第二種優選實施方式,優選利用3-溴-9,9』螺雙芴和硼酸衍生物官能化的化合物如2-硼酸苯並菲反應,在體系中加入四(三苯基膦)鈀、無水碳酸鉀、四氫呋喃和水,在氬氣保護下50-80℃下反應30-35小時,反應完畢。後處理同第一種優選實施方式。最終產品收率可達到94%。
如上所述的第三種優選實施方式,優選利用3-溴-9,9』螺雙芴在四氫呋喃中溶解,低溫下滴加正丁基鋰,在-78℃攪拌1小時,然後,將溶解在80ml中的氰基官能化的化合物如2-氰基苯並菲慢慢滴加到反應瓶中,1小時後自動升溫,反應過夜。在反應瓶中加入少量水淬滅反應,減壓蒸去溶劑,加入稀鹽酸,室溫攪拌,加入二氯甲烷萃取,分層,經過洗滌乾燥後可得產品,收率可達78%。
本發明還涉及本發明化合物在電子器件中、特別是在有機電致發光器件、有機場效應電晶體、有機太陽能電池中的用途。
本發明另外還涉及包含至少一種本發明化合物的電子器件。所述電子器件優選地選自有機電致發光器件(有機發光二極體、oled)、有機場效應電晶體(o-fet)、有機太陽能電池(o-sc)、有機薄膜電晶體(o-tft)、有機發光電晶體(o-let)、有機集成電路(o-ic)、有機染料敏化太陽能電池(odssc)、有機光學檢測器、有機光感受器、有機場猝熄器件(o-fqd)、發光電化學電池(lec)、有機雷射二極體(o-laser)和有機等離子體發射器件等,優選有機電致發光器件(oled)。
一個oled總體上包括:基板,例如(但不限於)玻璃、塑料、金屬;陽極,例如氧化銦錫(ito)陽極;空穴注入層(hil);空穴傳輸層(htl);電子阻擋層(ebl);有機發光層(eml);雷射阻擋層(ebl);電子傳輸層(etl);電子注入層(eil),例如lif、cs2co3;陰極,例如al。然而,應該指出,中間每層可以有一層或多層並且每層並非必須存在。
本發明的化合物可以用於該器件的任意一層或多層,但優選用於發光層(eml),更優選為發光層的主體材料,因為其具有高的三線態能級,穩定性好。本發明的化合物也可以用於作為激子阻擋材料或者電子傳輸材料用於激子阻擋層(ebl)和電子傳輸層(etl)中,因其載流子傳輸能力好。
本發明的有機發光層摻雜劑並無特別限定,優選為發光金屬配合物和發光有機分子,發光金屬配合物優選為銥和鉑的配合物,發光有機分子優選螢光化合物和熱激發延遲螢光化合物。為了提高器件效能,摻雜劑質量比例在6%-30%,優選6%-20%,進一步優選6%-15%。
在本發明的優選實施方式中,oled包括:基片/陽極/空穴注入材料(hil1)/空穴傳輸材料1(hil1)/空穴傳輸材料2(hil2)/電子阻擋層(ebl)/有機發光層(eml)/激子阻擋層(ebl)/電子傳輸層(etl)/陰極。其中,基片使用玻璃基板,ito作陽極材料。空穴注入材料使用現有技術中常用的2,3,6,7,10,11-六氰基-1,4,5,8,9,12-六氮雜苯並菲(hat-cn),空穴傳輸材料1使用現有技術中常用的n,n'-二苯基-n,n'-(1-萘基)-1,1'-聯苯-4,4'-二胺(npb);空穴傳輸材料2使用現有技術中常用的9,9',9」-三苯基-9h,9'h,9」h-3,3':6',3」-三咔唑(tris-pcz);電子阻擋層使用現有技術中常用的3,3'-二(n-咔唑基)-1,1'-聯苯(mcbp);有機發光層使用本發明所述化合物摻雜現有技術中常用的環金屬銥配合物ir(ppy)3、熱激發延遲螢光材料4czipn或藍色螢光材料bd1;激子阻擋層使用本發明所述化合物。另外,本發明所述化合物摻雜8-羥基喹啉-鋰或者金屬鋰也可用作電子傳輸層,部分器件也使用現有技術中常用的雙(10-羥基苯並[h]喹啉)鈹(be(bq)2)和三(8-羥基喹啉)鋁(alq3)。陰極採用金屬及其混合物結構,如mg:ag、ca:ag等,也可以是電子注入層/金屬層結構,如lif/al、liq/al和li2o/al。本發明優選電子注入材料為liq,陰極材料為al。
以下列舉實施例來說明本發明,本領域技術人員能夠理解,該例子僅是示例性的說明,並非窮盡性的說明。製備例為化合物合成例,所涉及的化學原料和試劑均為市售或按公開文獻合成,實施例為發光器件oled的製備。
製備例
中間體1的合成:
中間體1的結構式和合成路線如下式所示:
式m1化合物的製備方法為:在裝有溫度計、滴液漏鬥和機械攪拌的250ml三口瓶中,加入10.0g(36.21mmol)2-氨基-4』-溴-二苯甲酮及80ml水,在攪拌下慢慢滴加5.0g,98%(50mmol)濃硫酸,攪拌30分鐘,冷卻到0℃,滴加1.5g(21mmol)亞硝酸鈉溶於5ml水的溶液,滴加完畢,保持這個溫度30分鐘後,慢慢升溫到60℃,待氣泡完全放出後,冷卻到室溫,過濾,得到淡黃色結晶狀固體。固體經柱層析分離(350目矽膠,淋洗液為石油醚:二氯甲烷=20:1(v/v)),蒸去溶劑,乾燥後,得到淡黃色結晶7.50g,收率80%。ms(ei):m/z257.96[m+]。元素分析計算值c13h7bro(%):c60.26,h2.72;實測值:c60.23,h2.82。
式m2化合物的製備方法為:在200mlschlenk瓶中,在氬氣體保護下,加入2-溴聯苯4.6g(19.73mmol)及四氫呋喃80ml,溶解,冷卻到-78℃,在這個溫度下滴加2.4m,8.63ml(20.7mmol)正丁基鋰,在-78℃攪拌1小時,然後將溶解在80ml中的5.1g(19.73mmol)3-溴芴酮慢慢滴加到反應瓶中,1小時後自動升溫,反應過夜。在反應瓶中加入5ml水淬滅反應,並轉移到單口瓶中,在減壓下,用旋轉蒸發儀蒸去溶劑,分別加入80ml二氯甲烷和80ml水,溶解殘留物,並轉移到分液漏鬥中,分層,水層用25ml二氯甲烷萃取兩次,有機層再用80ml水洗兩次至中性。有機層用15g無水硫酸鈉乾燥3小時後,減壓蒸去溶劑二氯甲烷。將所得中間體3-溴-9-(2-二苯基)-9-芴醇直接投入到100ml單口瓶中,加入45ml乙酸,及4.5ml36%的濃鹽酸,115℃回流反應4小時,冷卻至室溫,過濾,得白色結晶。用10ml石油醚淋洗一次,乾燥後得5.2g,收率67%。ms(ei):m/z394.04[m+]。元素分析計算值c25h15br(%):c75.96,h3.82;實測值:c76.08,h3.75。
所述中間體1的製備方法為:在200mlschlenk反應瓶中,在氬氣保護下,投入3-溴-9,9』螺雙芴2.0g(5.05mmol)及80ml四氫呋喃,溶解後冷卻,在-78℃下滴加2.4m正丁基鋰2.2ml(5.28mmol),滴加完畢攪拌1小時,滴加硼酸三異丙酯1.42g(7.57mmol),攪拌1小時後,自動升溫,反應過夜。加入20ml氯化銨水溶液,淬滅反應。將反應物料轉移到單口瓶中,減壓蒸出溶劑後,在瓶中加入80ml二氯甲烷和80ml水,溶解,水洗,分層,水層用15ml二氯甲烷萃取兩次,合併有機相,再用80ml水水洗兩次,至中性。分去水層,有機層加15g無水硫酸鈉,乾燥3小時。減壓蒸去溶劑,得白色固體1.46g,收率80%。ms(ei):m/z360.16[m+]。元素分析計算值c25h17bo2(%):c83.36,h4.76;實測值:c83.41,h4.84。
製備例1:化合物1-75的合成
化合物1-75的結構式及合成路線如下式所示:
在200mlschlenk瓶中,加入3-硼酸-9,9』螺雙芴1.50g(4.16mmol)、2,4-二氯-6-苯基-1,3,5-三嗪0.47g(2.08mmol)、四(三苯基膦)鈀0.26g(0.23mmol)、無水碳酸鉀1.77g(12.8mmol),四氫呋喃50ml,水8ml,在氬氣體保護下,70℃攪拌反應30小時,反應完畢。蒸去溶劑,用50ml二氯甲烷和50ml水溶解殘留物,水洗,分出有機層,水層用15ml二氯甲烷萃取兩次,合併有機層,用50ml水洗兩次至中性,蒸去溶劑後,殘留物經柱層析分離(矽膠為350目,淋洗液為石油醚:三氯甲烷=2.5:1(v/v)),蒸去溶劑,乾燥後,得到1.27g白色結晶,收率為78%。ms(ei):m/z785.23[m+]。元素分析計算值c59h35n3(%):c90.16,h4.49;實測值:c90.18,h4.41。
附圖1是產品1-75的紫外吸收光譜、紫外吸收光譜(uv-vis)、室溫螢光光譜(pl)以及低溫磷光光譜(phos)。紫外吸收光譜和室溫螢光光譜在二氯甲烷的稀溶液(1×10-5mol/l)中測得;低溫磷光光譜在2-甲基四氫呋喃溶液(1×10-3mol/l)中測得。
製備例2:化合物1-123的合成
化合物1-123的結構式及合成路線如下式所示:
在200mlschlenk瓶中,加入3-硼酸-9,9』螺雙芴1.50g(4.16mmol)、三聚氯氰0.25g(1.38mmol)、四(三苯基膦)鈀0.26g(0.23mmol)、無水碳酸鉀1.77g(12.8mmol)、四氫呋喃50ml、水8ml,在氬氣體保護下,70℃攪拌反應30小時,反應完畢。蒸去溶劑,用50ml二氯甲烷和50ml水溶解殘留物,水洗,分出有機層,水層用15ml二氯甲烷萃取兩次,合併有機層,用50ml水洗兩次至中性,蒸去溶劑後,殘留物經柱層析分離(矽膠為350目,淋洗液為石油醚:三氯甲烷=2.5:1(v/v)),蒸去溶劑,乾燥後,得到1.13g白色結晶,收率為68%。ms(ei):m/z1023.41[m+]。元素分析計算值c78h45n3(%):c91.46,h4.43;實測值:c90.51,h4.41。
製備例3:化合物1-112的合成
化合物1-112的結構式及合成路線如下式所示:
在200mlschlenk瓶中,加入3-溴-9,9』螺雙芴3.95g(10.00mmol)、2-硼酸苯並菲2.72g(10.00mmol)、四(三苯基膦)鈀0.26g(0.23mmol)、無水碳酸鉀2.76g(20.0mmol)、四氫呋喃50ml、水8ml,在氬氣體保護下,70℃攪拌反應30小時,反應完畢。蒸去溶劑,用50ml二氯甲烷和50ml水溶解殘留物,水洗,分出有機層,水層用15ml二氯甲烷萃取兩次,合併有機層,用50ml水洗兩次至中性,蒸去溶劑後,殘留物經柱層析分離(矽膠為350目,淋洗液為石油醚:三氯甲烷=2.5:1(v/v)),蒸去溶劑,乾燥後,得到5.11g白色結晶,收率為94%。ms(ei):m/z542.21[m+]。元素分析計算值c43h26(%):c95.17,h4.83;實測值:95.19,h4.80。
製備例4:化合物1-113的合成
化合物1-113的結構式及合成路線如下式所示:
在200mlschlenk瓶中,在氬氣體保護下,加入3-溴-9,9』螺雙芴3.95g(10.00mmol)及四氫呋喃80ml,溶解,冷卻到-78℃,在這個溫度下,滴加2.4m,4.58ml(11.0mmol)正丁基鋰,在-78℃攪拌1小時,然後,將溶解在80ml中的2.53g(10.00mmol)2-氰基苯並菲慢慢滴加到反應瓶中,1小時後,自動升溫,反應過夜。在反應瓶中加入5ml水淬滅反應,並轉移到單口瓶中,在減壓下,用旋轉蒸發儀蒸去溶劑,加入10ml1m稀鹽酸,室溫攪拌2小時,加入30ml二氯甲烷萃取,分層,水層用25ml二氯甲烷萃取兩次,有機層再用30ml水洗兩次至中性。有機層用15g無水硫酸鈉乾燥3小時後,減壓蒸去溶劑二氯甲烷。蒸去溶劑後,殘留物經柱層析分離(矽膠為350目,淋洗液為石油醚:三氯甲烷=2.5:1(v/v)),蒸去溶劑,乾燥後,得到4.5g白色結晶,收率為78%。ms(ei):m/z570.20[m+]。元素分析計算值c44h26o(%):c92.60,h4.59;實測值:c92.64,h4.59。
製備例5:化合物1-1的合成
化合物1-1的結構式及合成路線如下式所示:
在200mlschlenk瓶中,加入3-硼酸-9,9』螺雙芴3.60g(10.00mmol)、1-溴-3,5-二苯基苯3.08g(10.00mmol)、四(三苯基膦)鈀0.26g(0.23mmol)、無水碳酸鉀2.76g(20.0mmol)、四氫呋喃50ml、水8ml,在氬氣體保護下,70℃攪拌反應30小時,反應完畢。蒸去溶劑,用50ml二氯甲烷和50ml水溶解殘留物,水洗,分出有機層,水層用15ml二氯甲烷萃取兩次,合併有機層,用50ml水洗兩次至中性,蒸去溶劑後,殘留物經柱層析分離(矽膠為350目,淋洗液為石油醚:三氯甲烷=2.5:1(v/v)),蒸去溶劑,乾燥後,得到4.82g白色結晶,收率為89%。ms(ei):m/z544.22[m+]。元素分析計算值c43h28(%):c94.82,h5.18;實測值:c94.60,h5.23。
製備例6:化合物1-17的合成
化合物1-17的結構式及合成路線如下式所示:
在200mlschlenk瓶中,加入3-硼酸-9,9』螺雙芴3.95g(10.00mmol)、1-溴-3,5-二(二苯並噻吩基)苯5.20g(10.00mmol)、四(三苯基膦)鈀0.26g(0.23mmol)、無水碳酸鉀2.76g(20.0mmol)、四氫呋喃50ml、水8ml,在氬氣體保護下,70℃攪拌反應30小時,反應完畢。蒸去溶劑,用50ml二氯甲烷和50ml水溶解殘留物,水洗,分出有機層,水層用15ml二氯甲烷萃取兩次,合併有機層,用50ml水洗兩次至中性,蒸去溶劑後,殘留物經柱層析分離(矽膠為350目,淋洗液為石油醚:三氯甲烷=2.5:1(v/v)),蒸去溶劑,乾燥後,得到6.24g白色結晶,收率為82%。ms(ei):m/z756.19[m+]。元素分析計算值c55h32s2(%):c87.27,h4.26;實測值:c87.25,h4.22。
實施例
器件製備:將塗布ito透明導電層的玻璃板在商用清洗劑中超聲處理,去離子水中衝洗,在丙酮和乙醇中各清洗三次,在潔淨的環境下烘烤至完全除去水分,用紫外光和臭氧清洗,並用低能陽離子束轟擊表面。把ito導電玻璃置入真空腔內,抽真空至2×10-5-2×10-5pa。然後在上述ito導電玻璃上依次蒸鍍空穴注入材料(hil1)、空穴傳輸材料1(hil1)、空穴傳輸材料2(hil2)、電子阻擋層(ebl)、有機發光層(eml)、激子阻擋層(ebl)、電子傳輸層(etl)以及陰極;其中,有機材料的蒸鍍速率為0.2nm/s,金屬電極的蒸鍍速率為0.5nm/s以上,發光層以雙源共蒸的方法,主體材料蒸鍍速率為0.2nm/s,摻雜材料的蒸鍍速率為0.03nm/s。
器件性能測試:器件的電流-亮度-電壓特性是由帶有校正過的矽光電二極體的keithley源測量系(keithley2400sourcemeter、keithley2000currentmeter)完成的,電致發光光譜是由photoresearch公司pr655光譜儀測量的,器件的外部量子效率通過文獻adv.mater.,2003,15,1043–1048的方法計算可得。以ir(ppy)3和4czipn作為摻雜劑的器件壽命是指以1000坎特拉每平方米為起始亮度,衰減到9000坎特拉每平方米(90%)的時間。而以bd1作為摻雜劑的器件壽命是指以5000坎特拉每平方米為起始亮度,衰減到4500坎特拉每平方米(90%)的時間。所有器件均在氮氣環境中封裝。
實施例涉及的化合物結構如下:
實施例(oled1-11)的結構和各層的膜厚厚度如下:
oled1:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-75:ir(ppy)36wt%(20nm)/1-75(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled2:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-75:4czipn15wt%(20nm)/1-75(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled3:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-75:bd110wt%(20nm)/1-75(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled4:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-75:ir(ppy)36wt%(20nm)/1-75(10nm)/1-75:20%liq(40nm)/liq(2nm)/al(100nm)
oled5:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-112:ir(ppy)36wt%(20nm)/1-75(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled6:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-112:4czipn15wt%(20nm)/1-75(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled7:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-113:ir(ppy)36wt%(20nm)/1-113(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled8:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-113:4czipn15wt%(20nm)/1-113(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled9:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-113:bd110wt%(20nm)/1-113(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled10:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-1:ir(ppy)36wt%(20nm)/host2(10nm)/balq(40nm)/liq(2nm)/al(100nm)
oled11:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/1-17:ir(ppy)36wt%(20nm)/1-75(10nm)/balq(40nm)/liq(2nm)/al(100nm)
比較例
比較例1和2的製備方法與實施例相同,僅是改變主體材料和激子阻擋材料化合物,其比較例1的器件結構如下:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/host1:bd110wt%(20nm)/host1(10nm)/balq(40nm)/liq(2nm)/al(100nm)
比較例2的器件結構如下:
ito/hat-cn(10nm)/npb(40nm)/mcbp(15nm)/host2:ir(ppy)36wt%(20nm)/host2(10nm)/balq(40nm)/liq(2nm)/al(100nm)
器件的性能數據見下表:
表1:器件性能數據
由表1可見,本發明的化合物獲得非常好的性能數據。比較例1和oled9均採用bd1作為摻雜劑,區別僅在於oled9的主體材料和激子阻擋材料為本發明的化合物1-113,通過器件性能數據的對比可見,oled9具有更低的工作電壓,外量子效率相對提高了將近3倍,器件壽命(90%)有顯著提高,從102小時變成625小時。另外,比較例2和oled1均採用ir(ppy)3作為摻雜劑,區別僅在於oled1的主體材料和激子阻擋材料為本發明的化合物1-75,通過器件性能數據的對比可見,oled1具有更長的器件壽命。附圖2是oled1-3的有機電致發光光譜,可以看出其具有很強的發光強度,其中oled3採用藍色螢光材料bd1為客體,其藍光峰值在455nm處且具有非常窄的半峰寬,證明其藍光的色純度較高。由此可見,本發明的化合物相對於現有技術常用的材料可以有效降低工作電壓、提高外量子效率和器件壽命。