利用1‑氰基環己基乙酸直接合成加巴噴丁的方法與流程
2023-09-11 11:04:50 3
(一)技術領域
本發明涉及一種加巴噴丁的合成工藝,即1-(氨甲基)環己基乙酸的合成工藝,特別涉及一種用1-氰基環己基乙酸直接合成加巴噴丁的方法,屬於藥物合成的技術領域。
(二)
背景技術:
加巴噴丁(gabapentin),商品名為neurontin,化學名為1-(氨甲基)環己基乙酸,結構與γ-氨基丁酸(gaba)類似,最早由美國的warner-lambert公司研製開發,並於1993年首次在英國上市。加巴噴丁是新一代的抗癲癇藥物,同時也適用於各種類型的神經痛,具有不良反應輕微且持續時間短,不經肝代謝等特點。自其上市以來,銷售額連續上升,在抗癲癇藥市場中佔有較大份額。
由於加巴噴丁含有一個環己烷基的結構,因此,在現有的報導中加巴噴丁的合成多以環己烷化合物為起始原料,其他的還有苯甲腈等。然而目前,這些途徑多為化學合成途徑,均存在一定的缺陷。
在us20030009055中以環己酮為原料,在氫氧化鉀,乙腈中55℃加熱回流7h,加水溶解,分層乾燥後得到環己基乙腈。環己基乙腈在含有dmso,碳酸鉀和硝基甲烷的環境中加熱,獲得1-硝甲基環己基乙腈,溶於乙醇,通過pd/c催化加氫,得到3-氨基-2-氮雜螺[4.5]癸烷-2-醇。之後在氫氧化鈉溶液中加熱回流,加入鹽酸調節ph,低溫析出2-羥基-2-氮雜螺[4.5]癸烷-3-酮,後溶於甲醇,通過雷尼鎳催化加氫,乾燥後,在鹽酸中加熱回流,冷卻後用二氯甲烷萃取乾燥。經離子交換獲得加巴噴丁。該方法需要使用具有一定毒性和爆炸性的硝基甲烷,而且需要經過兩次催化加氫對設備要求較高。
在us68469508中,利用1,1-環己基-二乙酸為原料,在乙酸酐和乙酸銨的混合液中,高溫反應,調節不同的ph值,在利用叔丁醇萃取,之後與氫氧化鈉/次氯酸鈉反應,鹽酸水解,再經過二環乙胺鹼化,甲醇、異丙醇重結晶得到加巴噴丁精品。其中二環乙胺處理後可用氫氧化鈉吸附氯離子回收二環乙胺循環利用。該法採用乙酸酐和乙酸銨的混合物,一步得到內醯胺,而且對二環乙胺進行了回收再利用,但整體收率較低,其中的一些步驟對設備的要求高,工藝仍較為繁瑣。
在ep0414262和us5132451中1-氰基環己基乙腈在乙醇,甲苯,氯化氫的環境下反應,通過移除多餘氣體,調節ph,水洗,再以甲苯作為相轉移試劑,後與甲醇,氫氧化鈉溶液反應,調節ph,低溫結晶得到1-氰基環己基乙酸。1-氰基環己基乙酸在鹼性條件下,通過雷尼鎳催化加氫,加入冰乙酸調節ph至中性,以四氫呋喃結晶得到加巴噴丁粗品,再以甲醇、水和異丙醇重結晶得到加巴噴丁精品。該方法的原料使用量大,廢物排量大,收率較低。
在上述路徑中由於其環境友好性差(消耗大量的hcl氣體,甲醇,甲苯,氫氧化鈉,水等試劑),收率低(收率最高達61.62%),經濟性較低,因而限制了其工業化應用。因此,在此路線上我們對其進行了改進。由於生物催化則具有反應條件溫和,具有良好的選擇性,催化效率高等優點。因此需要篩選出能夠特異選擇性催化1-氰基環己基乙腈且高酶活的腈水解酶。
此前在本實驗室發表的一篇論文中報導(processbiochemistry49(2014),2141-2148)利用純酶催化1-氰基環己基乙腈生產1-氰基環己基乙酸。然而純酶催化存在明顯的弊端:穩定性差、需要繁瑣複雜的純化過程、酶催化劑不易回收等等。因此,為了適用於工業化生產,我們決定利用含腈水解酶的靜息細胞催化1-氰基環己基乙腈生產1-氰基環己基乙酸,這在我們實驗室發表的另外的一項專利(cn104212850a)和論文(catalysiscommunications66(2015),121-125)均有詳細的報導。
在利用含腈水解酶的靜息細胞催化1-氰基環己基乙腈生成1-氰基環己基乙酸的過程中,由於靜息細胞的破碎,導致大量的蛋白質及有機大分子釋放到胞外,這對於下一步的催化加氫反應是不利的,這是由於蛋白質等大分子物質會使得加氫催化劑中毒。此外利用靜息細胞作為催化劑催化1-氰基環己基乙腈時,在經過一批反應後,酶活力迅速衰減,無法重複使用。因此經過後續改進,採用固定化技術對細胞進行固定化,這在本實驗室發表的另外一項專利(cn10491225a)中有詳細的報導。採用固定化技術可以極大的增強酶的穩定性,增強機械強度,保證細胞的完整性,重複利用等。
在xue等人發表的論文和專利cn10491225a中均有報導利用由靜息細胞或者固定化細胞催化1-氰基環己基乙腈合成關鍵中間體1-氰基環己基乙酸,通過雷尼鎳催化加氫合成加巴噴丁內醯胺。加巴噴丁內醯胺在鹽酸的作用下水解,碳酸鈉溶液調節ph,中和過多的氯離子,經異丙醇重結晶後得到加巴噴丁精品。該方法以酶法催化和化學催化相結合,反應條件溫和,特異選擇性高,環境友好,整體收率較高,步驟簡短。
目前,多數學者採用化學法合成加巴噴丁,需要使用大量的試劑,條件步驟冗長、繁瑣;而之前xue等人採用化學-酶法合成加巴噴丁需要通過酶法催化,直接加氫和化學水解三個主要步驟,步驟仍較為繁瑣。
因此通過對比研究發現,前人對於直接一步加氫催化合成加巴噴丁並未有大量研究,而且之前採用的工藝是將1-氰基環己基乙酸直接加氫合成環化物,再水解得到加巴噴丁。因此研究開發一種只經過一步加氫技能合成加巴噴丁的技術具有極大的意義。此外,需要設計產品的提純工藝和中間產物的循環利用工藝,從而實現完全轉化。本發明正是對於一步加氫催化合成加巴噴丁以及加巴噴丁的提純和1-氰基環己基乙酸的循環利用這一思路展開了研究。
(三)
技術實現要素:
本發明目的是提供一種通過1-氰基環己基乙酸的直接催化加氫生成加巴噴丁的製備方法,即通過對含有1-氰基環己基乙酸的酶催化轉化液進行直接加氫生產加巴噴丁,以及通過離子交換分離加氫轉化液中的加巴噴丁和1-氰基環己基乙酸,並對分離出來的1-氰基環己基乙酸進行循環利用的方法。
本發明採用的技術方案是:
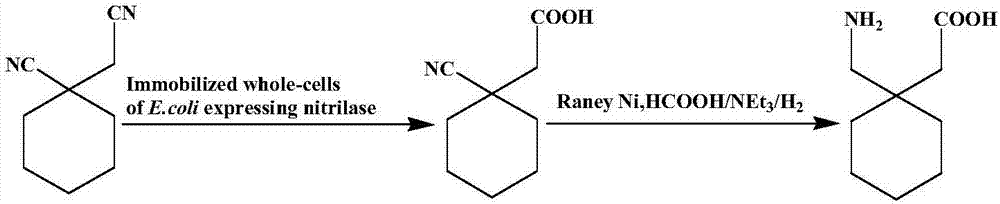
本發明提供一種利用1-氰基環己基乙酸直接合成加巴噴丁的方法(工藝流程如圖1),所述方法為:(1)轉化液:將腈水解酶基因工程菌固定化細胞分散於去離子水中,加入1-氰基環己基乙腈,在20-50℃、10-350rpm條件下攪拌反應完全後(優選35℃,200rpm下攪拌反應8h),抽濾,獲得濾餅a和含1-氰基環己基乙酸的濾液a;濾餅a為回收固定化細胞;所述腈水解酶基因工程菌固定化細胞是將seqidno.1所示腈水解酶基因構建的工程菌以硅藻土為載體製備獲得的(具體製備方法參照專利申請cn104911225a實施例1,腈水解酶基因同cn104911225a中seqidno.1所示);所述1-氰基環己基乙腈與固定化細胞質量比為1:1.0-1.1,所述去離子水的體積用量以1-氰基環己基乙腈質量計為6.5-7.0ml/g;(2)加巴噴丁的合成:取步驟(1)濾液a至加氫反應釜中,加入催化劑和助劑,通入氫氣,於20-150℃、300-1100rpm條件下反應(優選以10℃幅度逐步升溫模式進行反應,每次升溫後通入氫氣反應12h,再排除氫氣,優選反應溫度為30-80℃),反應完全後將反應液冷卻至30-80℃(優選60℃),排空氫氣,過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑;所述催化劑為雷尼鎳、銠碳(銠質量負載量5%)或鈀炭(鈀質量負載量10%)(優選雷尼鎳),所述助劑有機酸、無機酸、無機鹼或有機鹼,更優選為甲酸、乙酸、甲酸銨或三乙胺中的一種或多種;所述催化劑與濾液a中1-氰基環己基乙酸質量比為0.1-1:1,所述助劑用量以濾液a體積計為0.5-2.5%(優選1-2%);(3)分離純化:將步驟(2)獲得的濾液b冷卻後調節ph值至4.0-5.0,上樣過陽離子交換樹脂,首先以蒸餾水洗滌,收集ph為3.0-6.0範圍內的流出液a為含有1-氰基環己基乙酸的洗出液;再以體積濃度15%的氨水溶液為洗脫液,收集ph為7.5-13.0範圍內的流出液b;流出液b減壓濃縮至原體積的1/10,獲得濃縮液a,濃縮液a加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至原體積的1/6-1/5,獲得的濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,收集晶體乾燥,得到加巴噴丁;流出液a回收循環利用。
進一步,步驟(2)所述助劑為甲酸和三乙胺以體積比0.1-3.6:1的混合或者2m乙酸水溶液與三乙胺以體積比0.1-3.6:1的混合。
進一步,步驟(3)所述陽離子交換樹脂為001×7型強酸性陽離子交換樹脂,購買自寧波爭光樹脂廠,其他型號的陽離子交換樹脂也在本發明的保護範圍內。
進一步,步驟(2)所述反應以升溫幅度10℃的梯度逐步升溫至80℃,每次升溫後通入氫氣反應12h,然後排出氣體,通入氫氣量為0-2mpa(優選0.5-1.0mpa)。
進一步,步驟(2)所述反應在40℃,500rpm下反應12h後排除氫氣,升溫至50℃後再通入氫氣,保留12h後排除氫氣,升溫至60℃通入氫氣反應12h,排除氫氣後,將反應液過濾,獲得濾液b和濾餅b。
進一步,步驟(3)所述流出液b在50℃下濃縮至原來體積的1/10,獲得的濃縮液a加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至原體積的1/6-1/5,獲得的濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,有白色晶體析出,抽濾後濾餅烘乾即為加巴噴丁。
本發明所述催化劑銠碳的銠質量負載量為5%;鈀炭中鈀質量負載量為10%。所述雷尼鎳購買自大連通用化工有限公司,其他型號的雷尼鎳也在本發明的保護範圍內;鈀炭(鈀負載量為10%)購買自上海麥克林生化科技有限公司,其他型號的鈀炭也在本發明的保護範圍內。
本發明步驟(3)中流出液a(即含1-氰基環己基乙酸的溶液)的循環利用可採用直接加氫反應,也可採用與步驟(1)的濾液a(即固定化細胞催化後的1-氰基環己基乙酸轉化液)混合後再進行直接加氫反應,更優選採用與固定化細胞催化的1-氰基環己基乙酸轉化液混合後再加氫。
本發明所述濾液a和濾液b,濾餅a和濾餅b均為不同步驟過濾所得濾液和濾餅,所述流出液a和流出液b為不同洗脫液獲得的流出液,濃縮液a和濃縮液b均指濃縮液,字母本身沒有含義。
與現有技術相比,本發明有益效果主要體現在:
原子經濟性好,採用生物催化與化學催化相結合生產加巴噴丁,其中在催化加氫步驟中能夠將固定化細胞催化後的反應液直接用於加氫反應生成產物加巴噴丁,因此避免提純1-氰基環己基乙酸,且利用水作為介質,利於回收循環利用,減少汙染。而且通過助劑的添加和升溫策略的改變,可直接催化1-氰基環己基乙酸一步合成加巴噴丁,在最優條件下,反應一個批次加巴噴丁收率達53.3%;利用離子交換法對加巴噴丁與1-氰基環己基乙酸進行分離,可將未反應完的1-氰基環己基乙酸回收循環利用於二次加氫反應,實現中間產物的完全轉化,通過過對1-氰基環己基乙酸的回收並進行加氫反應,經過5次循環後,使得加巴噴丁收率達到80%以上,比已報導的化學工藝提高了近20%(ep0414262)。
(四)附圖說明
圖1從1-氰基環己基乙腈選擇性水解、直接加氫一步合成加巴噴丁的路徑圖。
圖2為化學-酶法合成加巴噴丁的工藝流程圖。
圖3為不同的升溫策略對於1-氰基環己基乙酸在該體系中直接加氫生成加巴噴丁的反應曲線圖,a:實施例2梯度升溫;b:實施例8恆溫60℃。
圖4為1-氰基環己基乙酸經過回收循環利用後多批次反應柱狀圖。
(五)具體實施方式
下面結合具體實施例對本發明進行進一步描述,但本發明的保護範圍並不僅限於此:
lb液體培養基:酵母粉5g/l,蛋白腖10g/l,nacl10g/l,溶劑為去離子水,ph值自然。
發酵培養基:蛋白腖15g/l,酵母粉12g/l,nacl10g/l,甘油12g/l,(nh4)2so45g/l,kh2po41.36g/l,k2hpo4·3h2o2.28g/l,mgso4·7h2o0.375g/l,溶劑為去離子水,ph值自然。
加巴噴丁與1-氰基環己基乙酸的hplc檢測條件:色譜柱welchromc18柱(5μm×250mm×4.6mm);流動相:緩衝液(5mmnh4h2po4/13mmnaclo4·h2o,高氯酸調節ph至1.8):乙腈=76:24,v/v;柱溫:40℃;波長:215nm;流速1ml/min。
實施例1:一種化學-酶法合成加巴噴丁的方法,步驟如下:
(1)種子液的製備:將實驗室之前已構建好的含腈水解酶基因(seqidno.1)的工程菌e.colibl21(de3)/pet28b(+)-f168v(同專利申請cn104911225a實施例1)接種於含有終濃度5g/l卡那黴素的lb平板培養基,於37℃條件下過夜培養,挑取單菌落接種於100ml含有終濃度5g/l卡那黴素的lb液體培養基中,與37℃,150rpm下過夜培養,獲得種子液。
(2)發酵培養:接種種子液於含有終濃度5g/l卡那黴素的3l發酵培養基的5l發酵罐中,接種量為3%(v/v),調節通氣比為1.3vvm,在37℃,500rpm條件下進行發酵培養,發酵過程流加體積濃度8%的氨水和體積濃度10%的磷酸水溶液以維持發酵液ph在6.5左右,培養4h後,降溫至28℃,加入乳糖誘導,乳糖終濃度為12.5g/l(m/v),培養10h,停止發酵,放罐獲得發酵液。於8000rpm離心10min,棄上清,收集獲得溼菌體。
(3)固定化細胞的製備:取20g溼菌體完全懸浮於200ml蒸餾水中,添加1.2g硅藻土在磁力攪拌器攪拌1h,後加入6ml的體積濃度5%的聚乙烯亞胺水溶液攪拌1h,添加2ml的體積濃度25%戊二醛水溶液攪拌30min後抽濾,蒸餾水洗2-3次,濾餅即為固定化細胞,稱重得31.44g。
(4)中間產物1-氰基環己基乙酸的合成:取15.72g固定化細胞於100ml去離子水中,攪拌使其分散均勻後加入14.8g1-氰基環己基乙腈,在35℃,200rpm下攪拌反應8h後,抽濾,獲得濾液a和濾餅a。濾液a(hplc檢測1-氰基環己基乙酸濃度為1mm)用於下一步的催化加氫,濾餅a為固定化細胞,用於下一個批次的酶催化反應。
(5)加巴噴丁的合成:取用蒸餾水稀釋4倍後的步驟(4)濾液a(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜中,加入約1.0graneyni(購自大連通用化工有限公司),加入1ml(98%)三乙胺和0.5ml(98%)甲酸;通入氮氣置換空氣,置換3次後,通入1mpa氫氣於40℃,500rpm下保留12h後排除氫氣,升溫至50℃後再通入氫氣,保留12h後排除氫氣後,升溫至60℃通入氫氣後,保留12h。排除反應釜內的氣體後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。
(6)加巴噴丁的提純:取上述濾液b加入提前配製好的2mol/l甲酸水溶液調節ph至5.0左右;通過001×7型強酸性陽離子交換樹脂(購買自寧波爭光樹脂廠)吸附,完全吸附之後首先用蒸餾水洗滌,收集ph值在3.0-6.0範圍內的流出液a,再用體積濃度為15%的氨水洗脫,收集ph值在7.5-13.0範圍內的流出液b(即獲得含有加巴噴丁的洗脫液)。流出液b200ml在50℃下濃縮至20ml,獲得濃縮液a,再加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至10ml,獲得的濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,有白色晶體析出,抽濾後濾餅烘乾即為加巴噴丁,質量收率達到50%。流出液a(即未吸附在陽離子交換樹脂上的物質)為含有1-氰基環己基乙酸的轉化液,回收用於二次加氫使用。
剩餘1-氰基環己基乙酸的循環利用:經過hplc檢測,流出液a中1-氰基環己基乙酸未發生較大改變以及未引入其他雜質。並與上述固定化細胞催化轉化液(即步驟(4)濾液a)混合後濃度為215.6mm,按照上述加巴噴丁合成體系進行二次加氫。在對1-氰基環己基乙酸進行5次循環利用之後,加巴噴丁收率達到80%以上(1-氰基環己基乙酸的回收利用如圖4)。
實驗例2:含1-氰基環己基乙酸的固定化細胞催化的轉化液直接加氫生產加巴噴丁
(1)加巴噴丁的合成:取實施例1中步驟(4)的含1-氰基環己基乙酸的固定化細胞催化的濾液a用蒸餾水稀釋4倍後(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜中,加入約1.0graneyni,加入1ml98%三乙胺和0.5ml98%甲酸;通入氮氣置換空氣,置換3次後,通入1mpa氫氣於30℃,500rpm下保留12h後排除氫氣,升溫至40℃,500rpm下保留12h後排除氫氣,升溫至50℃後再通入氫氣,保留12h後排除氫氣後,升溫至60℃通入氫氣後,保留12h,通入氫氣於70℃,500rpm下保留12h後排除氫氣,升溫至80℃後再通入氫氣,保留12h後排除氫氣後,反應液降溫至60℃後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。反應進程見圖3中a所示。
(2)加巴噴丁的提純:取上述濾液b加入提前配製好的2mol/l甲酸水溶液調節ph至5.0左右;通過001×7型強酸性陽離子交換樹脂(購買自寧波爭光樹脂廠)吸附,完全吸附之後首先用蒸餾水洗滌,收集ph值在3.0-6.0範圍內的流出液a,再用體積濃度為15%的氨水洗脫,收集ph值在7.5-13.0範圍內的流出液b(即獲得含有加巴噴丁的洗脫液)。流出液b200ml在50℃下濃縮至20ml,獲得的濃縮液a加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至10ml,獲得的濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,有白色晶體析出,抽濾後濾餅烘乾即為加巴噴丁,質量收率達到48%。流出液a(即未吸附在陽離子交換樹脂上的物質)為含有1-氰基環己基乙酸的轉化液,回收用於二次加氫使用。
實驗例3:含1-氰基環己基乙酸的固定化細胞催化的轉化液直接加氫生產加巴噴丁
(1)加巴噴丁的合成:取實施例1步驟(4)中的含1-氰基環己基乙酸的固定化細胞催化的濾液a用蒸餾水稀釋4倍後(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜中,加入約1.0gpd/c(pd質量負載量10%),1ml98%三乙胺和0.5ml98%甲酸;通入氮氣置換空氣,置換3次後,通入1mpa氫氣於40℃,500rpm下保留12h後排除氫氣,升溫至50℃後再通入氫氣,保留12h後排除氫氣後,升溫至60℃通入氫氣後,保留12h。排氣後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。
(2)加巴噴丁的提純:取上述濾液b加入提前配製好的2mol/l甲酸水溶液調節ph至5.0左右;通過001×7型強酸性陽離子交換樹脂(購買自寧波爭光樹脂廠)吸附,完全吸附之後首先用蒸餾水洗滌,收集ph值在3.0-6.0範圍內的流出液a,再用體積濃度為15%的氨水洗脫,收集ph值在7.5-13.0範圍內的流出液b(即獲得含有加巴噴丁的洗脫液)。流出液b300ml在50℃下濃縮至30ml,獲得濃縮液a,取濃縮液a加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至15ml,將獲得的濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,有白色晶體析出,抽濾後濾餅烘乾即為加巴噴丁,質量收率達到84%,但同時約有10%加巴噴丁內醯胺。流出液a(即未吸附在陽離子交換樹脂上的溶液)為含有1-氰基環己基乙酸的轉化液,回收用於二次加氫使用。
結果表明,pd/c具有更佳的催化活力,但同時催化選擇性略低於raneyni。
實驗例4:含1-氰基環己基乙酸的固定化細胞催化的轉化液直接加氫生產加巴噴丁。
(1)加巴噴丁的合成:取實施例1步驟(4)中的含1-氰基環己基乙酸的固定化細胞催化的濾液a用蒸餾水稀釋4倍後(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜中,加入約1.0graneyni,加入1.07ml98%三乙胺和0.43ml98%甲酸;通入氮氣置換空氣,置換3次後,通入1mpa氫氣於40℃,500rpm下保留12h後排除氫氣,升溫至50℃後再通入氫氣,保留12h後排除氫氣後,升溫至60℃通入氫氣後,保留12h,排氣後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。
(2)加巴噴丁的提純:取上述濾液b加入提前配製好的2mol/l甲酸水溶液調節ph至5.0左右;通過001×7型強酸性陽離子交換樹脂(購買自寧波爭光樹脂廠)吸附,完全吸附之後首先用蒸餾水洗滌,收集ph值在3.0-6.0範圍內的流出液a,再用體積濃度為15%的氨水洗脫,收集ph值在7.5-13.0範圍內的流出液b(即獲得含有加巴噴丁的洗脫液)。流出液b200ml在50℃下濃縮至20ml,獲得濃縮液a,將濃縮液a加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至10ml,將獲得的濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,有白色晶體析出,抽濾後濾餅烘乾即為加巴噴丁,質量收率達到60%。流出液a(即未吸附在陽離子交換樹脂上的溶液)為含有1-氰基環己基乙酸的轉化液,回收用於二次加氫使用。
結果表明,助劑中酸和鹼的比例影響1-氰基環己基乙酸在該體系中加巴噴丁的合成,最合適體積比例為1:2。
實施例5:含1-氰基環己基乙酸的固定化細胞催化的轉化液直接加氫生產加巴噴丁。
(1)加巴噴丁的合成:取實施例1中步驟(4)的含1-氰基環己基乙酸的固定化細胞催化的濾液a用蒸餾水稀釋4倍後(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜中,加入約1.0gpd/c(pd質量負載量10%),加入1ml98%三乙胺和0.5ml甲酸。通入氮氣置換空氣,置換3次,不通入氫氣,直接在40℃,500rpm下保留12h,升溫至50℃保留12h,後再升溫至60℃保留12h,排氣後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。
(2)加巴噴丁的提純:取上述濾液b加入提前配製好的2mol/l甲酸水溶液調節ph至5.0左右;通過001×7型強酸性陽離子交換樹脂(購買自寧波爭光樹脂廠)吸附,完全吸附之後首先用蒸餾水洗滌,收集ph值在3.0-6.0範圍內的流出液a,再用體積濃度為15%的氨水洗脫,收集ph值在7.5-13.0範圍內的流出液b(即獲得含有加巴噴丁的洗脫液)。流出液b200ml在50℃下濃縮至20ml,獲得濃縮液a,再加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至10ml,獲得的濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,有白色晶體析出,抽濾後濾餅烘乾即為加巴噴丁,質量收率達到37.2%。流出液a(即未吸附在陽離子交換樹脂上的物質)為含有1-氰基環己基乙酸的轉化液,回收用於二次加氫使用。
結果表明,在該體系中,即使沒有氫氣作為氫供體,pd/c依然能夠催化部分1-氰基環己基乙酸加氫合成加巴噴丁,這是由於甲酸可以作為氫供體。
實施例6:含1-氰基環己基乙酸的固定化細胞催化轉化液直接加氫合成加巴噴丁
(1)加巴噴丁的合成:取實施例1中步驟(4)的含1-氰基環己基乙酸的固定化細胞催化的濾液a用蒸餾水稀釋4倍後(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜中,加入約1.0graneyni,加入0.715ml98%三乙胺和0.285ml甲酸;通入氮氣置換空氣,置換3次後,通入1mpa氫氣於40℃,500rpm下保留12h後排除氫氣,升溫至50℃後再通入氫氣,保留12h後排除氫氣後,升溫至60℃通入氫氣後,保留12h,排氣後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。
(2)加巴噴丁的提純:取上述濾液b加入提前配製好的2mol/l甲酸水溶液調節ph至5.0左右;通過001×7型強酸性陽離子交換樹脂(購買自寧波爭光樹脂廠)吸附,完全吸附之後首先用蒸餾水洗滌,收集ph值在3.0-6.0範圍內的流出液a,再用體積濃度為15%的氨水洗脫,收集ph值在7.5-13.0範圍內的流出液b(即獲得含有加巴噴丁的洗脫液)。流出液b200ml在50℃下濃縮至20ml,獲得的濃縮液a加入2倍體積的異丙醇靜置1h後再次在50℃下濃縮至10ml,獲得濃縮液b加入2-5倍體積的異丙醇冰浴攪拌後在0-4℃下冷卻靜置,有白色晶體析出,抽濾後濾餅烘乾即為加巴噴丁,質量收率達到50.6%。流出液a(即未吸附在陽離子交換樹脂上的物質)為含有1-氰基環己基乙酸的轉化液,回收用於二次加氫使用。
結果表明,減少甲酸和三乙胺的加入量,對於加巴噴丁的積累具有一定的抑制。
實施例7:含1-氰基環己基乙酸的固定化細胞催化轉化液直接加氫生成加巴噴丁
加巴噴丁的合成:取實施例1中步驟(4)的含1-氰基環己基乙酸的固定化細胞催化的濾液a用蒸餾水稀釋4倍後(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜中,加入約1.0graneyni,加入1.0ml98%三乙胺和3.5ml2m的乙酸水溶液;通入氮氣置換空氣,置換3次後,通入1mpa氫氣於40℃,500rpm下保留12h後排除氫氣,升溫至50℃後再通入氫氣,保留12h後排除氫氣後,升溫至60℃通入氫氣後,保留12h,排氣後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。對濾液b進行hplc檢測,加巴噴丁收率達到81.6%。
實施例8:含1-氰基環己基乙酸的固定化細胞催化轉化液直接加氫生成加巴噴丁
(1)加巴噴丁合成:取實施例1中步驟(4)含1-氰基環己基乙酸的固定化細胞催化的濾液a用蒸餾水稀釋4倍後(1-氰基環己基乙酸濃度為250mm)100ml至加氫反應釜,加入約1.0graneyni,加入0.715ml98%三乙胺和0.285ml甲酸;通入氮氣置換空氣,置換3次後,通入1mpa氫氣於60℃,500rpm下反應24h,排除氫氣後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b冷卻至室溫。利用hplc檢測濾液b,結果顯示反應液中只有少量的加巴噴丁生成,大部分產物為加巴噴丁內醯胺,底物轉化率只有61.5%。反應進程見圖3中b所示。
結果表明,在某一溫度下進行恆溫催化不利於加巴噴丁在該加氫體系中積累。
實施例9:含1-氰基環己基乙酸晶體直接加氫生成加巴噴丁
(1)1-氰基環己基乙酸晶體的提純:取實施例1中步驟(4)含1-氰基環己基乙酸的固定化細胞催化的濾液100ml(1-氰基環己基乙酸濃度為250mm)與0.6g活性炭混合後在60℃條件下攪拌1h,待混合液降溫至室溫後,抽濾,濾液用2mhcl溶液調節ph至2.0,此時有大量晶體析出,抽濾即可獲得1-氰基環己基乙酸晶體,在50℃過夜烘乾,即可獲得乾燥的1-氰基環己基乙酸晶體。
(2)加巴噴丁合成:取14.5g1-氰基環己基乙酸晶體溶於70ml甲醇,之後加入1grh/c(rh質量負載量為5%);通入氮氣置換空氣,置換3次後,通入0.5mpa氫氣於室溫下,反應2h,排氣後,將反應液過濾,獲得濾液b和濾餅b,濾餅b為回收催化劑,濾液b即為加氫反應液,經hplc檢測,發現只有少量的加巴噴丁生成,轉化率極低。
結果表明,rh/c在催化溶於甲醇的1-氰基環己基乙酸生成加巴噴丁時,催化活性低,無法達到工業化生產的要求。