一種矽碳複合負極材料的製備方法及鋰離子電池與流程
2023-12-03 04:07:01 2
本發明屬於鋰離子電池負極材料領域,尤其涉及一種首次庫侖效率高、循環性能好、壓實密度高、電極結構穩定的矽基複合負極材料的製備方法,以及使用該負極材料的鋰離子電池。
背景技術:
鋰離子電池以其比能量大、工作電壓高、循環使用壽命長、體積小、重量輕、綠色環保等優勢廣泛應用於各種可攜式電子設備和電動汽車中。目前商業化的鋰離子電池負極材料負極材料主要為石墨,包括天然石墨、人造石墨等,但其理論比容量僅為372mAh/g,已難以滿足鋰離子電池應用領域對高能量密度電源的需求。因此,開發新型高比容量的鋰離子電池負極材料已成為迫切的課題。
在非碳負極材料中,矽基材料由於具有最高的理論嵌鋰容量4200mAh/g,遠高於其它所有負極材料的理論嵌鋰容量,且矽的儲量豐富(地殼元素含量中排第二位),是非常具有潛力成為下一代鋰離子電池的負極材料,因此成為研究的熱點。然而,矽基材料在高程度脫嵌鋰的條件下,存在著高達300%以上的體積效應,由此產生的機械作用力會造成矽顆粒的破碎、粉化,使矽顆粒與集流體的電接觸喪失,造成矽負極材料容量的急劇衰減,表現為極差的循環穩定性。另外,矽是一種半導體材料,其本徵電導率僅為6.7×10-4S/cm。
針對上述問題,目前提出的改性方法中比較有效的是製備矽碳複合材料來緩解電池充放電過程中的體積膨脹,此方法已經廣泛應用於鋰離子電池負極材料的改性研究中。CN103531760公開的蛋黃-蛋殼結構多孔矽碳複合微球製備方法,其製備工序過於複雜,中空內徑控制過於困難,雖然能供矽的一定膨脹空間,但振實密度不高,導電性較差,且需採用氫氟酸刻蝕,對環境汙染嚴重;CN103000901公開的採用PVC包覆矽粉製備的無定形碳包覆矽顆粒的製備方法,雖然能在一定程度遏制住體積效應,但導電性較差,且PVC包覆層較脆,易被破壞,不利於長期循環。因此,開發一種高導電性、高容量、首次庫侖效率高、循環穩定性好的製備工藝,仍是目前矽基材料領域要解決的難題。
技術實現要素:
針對現有技術的不足,本發明的目的在於提出一種矽碳負極材料的製備方法及鋰離子電池,本發明製備的矽碳負極材料具有首次庫侖效率高、循環性能好、壓實密度高、電極結構穩定等優點,且該複合負極材料的製備過程環境友好無汙染。
為了實現上述目的,本發明採用以下技術方案:
一種矽碳複合負極材料的製備方法,將納米矽、石墨微粉置於球磨機中,在有機溶劑的環境中球磨均勻分散,真空乾燥後與瀝青置於錐形混合機中進行粗混,再將粗混後的混合粉末置於機械融合機內進行機械融合,最後在惰性氣體的保護下進行熱處理,冷卻後得到矽碳複合負極材料。在本技術方案中,對納米矽進行瀝青軟化包覆,可避免矽顆粒與電解液直接接觸,減緩容量衰減速度,同時縮短了鋰離子的擴散路徑,保證了電極材料的電子傳導不會喪失,即提高首次充放效率,充放電容量和循環性能;包覆前,先利用石墨微粉將納米矽分散,避免在與瀝青包覆時,納米矽聚集導致局部容量過剩,使得納米矽分散均勻。
採用機械融合可以改善顆粒表面狀態,減少顆粒表面活性點,提高循環性能,以及改善材料與電解液的相容性,減輕了充放電過程中體積膨脹的現象。
作為優選,所述矽碳複合負極材料中原料的質量分數分別為:納米矽10-40%,石墨微粉30-80%,瀝青10-30%。
作為優選,納米矽的中值粒徑為50-200nm;石墨微粉為鱗片狀石墨、球形石墨或人造石墨中的一種;所述石墨微粉的中值粒徑為5-15μm;所述有機溶劑為乙醇、丙酮、乙腈、四氫呋喃、三氯甲烷、N-甲基吡咯烷酮或N,N-二甲基甲醯胺中的一種;瀝青為低溫煤瀝青、中溫煤瀝青、高溫煤瀝青、渣油瀝青、焦油瀝青、天然瀝青、石墨瀝青或頁巖瀝青中的一種;所述瀝青的中值粒徑為1-20μm。
作為優選,球磨時間為3-10h,機械融合機的轉速為500-1000r/min,機械融合時間15-60min。
作為優選,熱處理為以0.5-5℃/min的升溫速率升至500℃,保溫1-200min,再以0.5-10℃/min的升溫速率升至900-1200℃,保溫10-240min,最後自然或程序降溫至室溫。
作為優選,納米矽在進行球磨前,先將矽粉添加至其8-12倍質量的濃度為8-12wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至110-130℃,保溫疏解6-8h;疏解後過濾得到的矽粉添加至其8-12倍質量的濃度為70-80wt%的乙醇溶液中,加熱至80-90℃後保溫3-5h,再次進行過濾,打漿至漿料叩解度為14-16°SR,然後進行磨漿,磨漿至漿料叩解度為18-22°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其18-22倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.4-0.6倍的海藻提取液,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為2-4wt%的氯化鈣溶液,在40-50℃下交聯1-2h後,轉移液氮中冰凍1-2天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽。
在本技術方案中,對納米矽進行改性,以使得後續與石墨微粉球磨時分散更均勻,可以提高碳矽複合負極材料與電解液的相容性。
作為優選,海藻提取液的固含量為25-35wt%。
作為優選,機械融合機為臥式融合機或立式融合機;惰性氣體包括氮氣、氬氣、氖氣、氦氣或疝氣中的一種。
一種鋰離子電池,負極材料為上述製備的矽碳複合負極材料。
作為優選,矽碳負極材料、導電劑和粘結劑按質量百分比80-94:3-10:5-10溶解在溶劑中混合,塗覆於銅箔集流體上,真空乾燥製得負極;導電劑為Super P-Li、乙炔黑、碳納米管、石墨烯、納米碳纖維、富勒烯的至少一種;粘結劑為聚醯亞胺樹脂、丙烯酸樹脂、聚偏二氟乙烯、聚乙烯醇、羧甲基纖維素鈉、丁苯橡膠或海藻酸鈉中的一種;溶劑為去離子水、N-甲基吡咯烷酮、二甲基甲醯胺、丙酮或甲基乙基酮中的一種。
本發明的有益效果是:
1)本發明對納米矽進行瀝青軟化包覆,可避免矽顆粒與電解液直接接觸,減緩容量衰減速度,同時縮短了鋰離子的擴散路徑,保證了電極材料的電子傳導不會喪失,即提高首次充放效率,充放電容量和循環性能;包覆前,先利用石墨微粉將納米矽分散,避免在與瀝青包覆時,納米矽聚集導致局部容量過剩,使得納米矽分散均勻;
2)採用機械融合可以改善顆粒表面狀態,減少顆粒表面活性點,提高循環性能,以及改善材料與電解液的相容性,減輕了充放電過程中體積膨脹的現象。
3)本發明生產效率高,節省成本,製備過程安全,製備過程環境友好無汙染,可用於工業化生產。
附圖說明
圖1為本發明實施例1製備的矽碳負極材料的XRD圖。
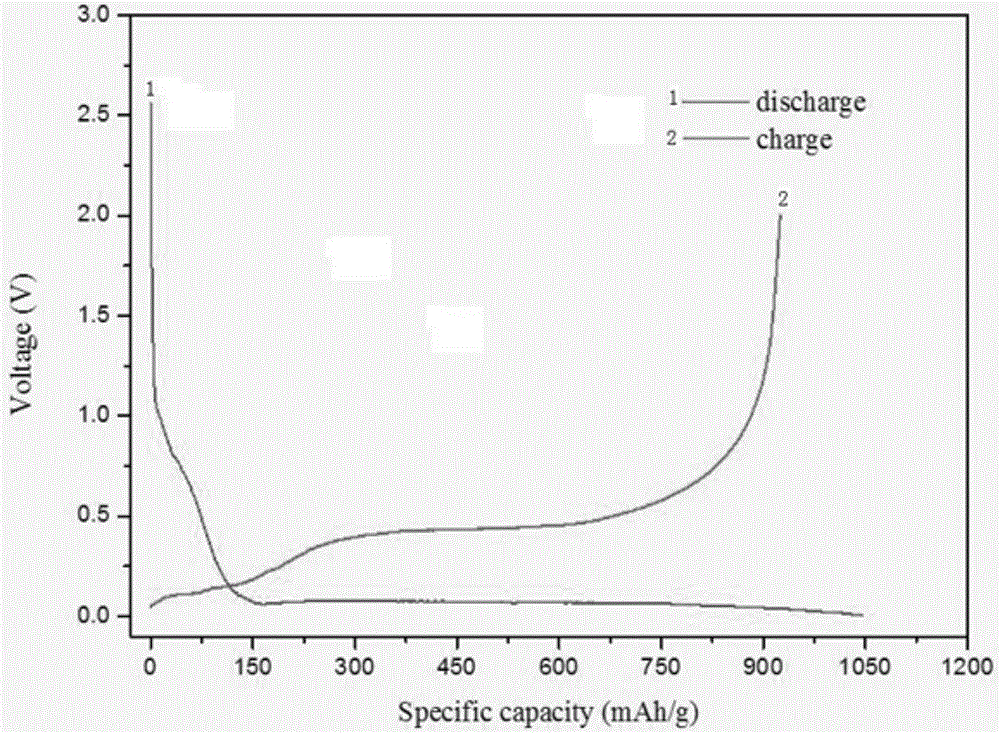
圖2為本發明實施例4製備的矽碳負極材料的首次電壓容量圖。
圖3位本發明實施例8製備的矽碳負極材料的循環性能曲線。
具體實施方式
下面通過具體實施例,對本發明的技術方案作進一步的具體說明。應當理解,本發明的實施並不局限於下面的實施例,對本發明所做的任何形式上的變通和/或改變都將落入本發明保護範圍。
在本發明中,若非特指,所有的份、百分比均為重量單位,所採用的設備和原料等均可從市場購得或是本領域常用的。下述實施例中的方法,如無特別說明,均為本領域的常規方法。
納米矽的中值粒徑為50-200nm;石墨微粉的中值粒徑為5-15μm;瀝青的中值粒徑為1-20μm;
實施例1:
一種矽碳複合負極材料的製備方法,包括以下步驟:
A:將120g球形石墨超聲分散於100ml無水乙醇中,然後將納米矽40g加入石墨分散液中,採用行星式球磨機球磨7h,球磨機轉速為250r/min,然後放置真空乾燥箱中80℃真空乾燥3h;
納米矽在進行球磨前,先將矽粉添加至其10倍質量的濃度為10wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至120℃,保溫疏解7h;疏解後過濾得到的矽粉添加至其10倍質量的濃度為75wt%的乙醇溶液中,加熱至85℃後保溫4h,再次進行過濾,打漿至漿料叩解度為15°SR,然後進行磨漿,磨漿至漿料叩解度為20°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其20倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.5倍的海藻提取液,海藻提取液的固含量為30wt%,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為3wt%的氯化鈣溶液,在45℃下交聯1.5h後,轉移液氮中冰凍2天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽;
B:將60g中溫煤瀝青與上述A中製備的矽/石墨複合物於錐形混合機中進行混合,混合時間為30min;然後將上述混合物置於臥式機械融合機內進行機械融合,機械融合機的轉速為800r/min,融合時間為25min;最後將融合後的混合物轉移至管式爐中高溫炭化處理,溫度程序為:室溫下以2℃/min的速率升溫至500℃,保溫30min,然後以5℃/min的速率升溫至1000℃,保溫180min,最後自然冷卻至室溫,粉碎過篩即可得到矽碳複合材料。
為了檢驗本發明鋰離子電池矽碳複合負極材料的性能,組裝成半電池對其進行性能測試。將所得的鋰離子電池矽碳複合負極材料分別與導電劑super P-Li、粘結劑CMC和SBR按照質量比80:10:10球磨混合,用去離子水調節混合物的粘度製成漿料,均勻塗覆在銅箔上,80℃真空乾燥8h,製得實驗電池用極片。再以鋰片作為對電極在手套箱中組裝成CR2032型扣式電池,首個循環採用0.05C倍率下進行活化,之後採用0.2C倍率充放電電壓範圍為0.01-1.5V進行循環性能測試,測試結果見圖1與表1。
實施例2:
一種矽碳複合負極材料的製備方法,包括以下步驟:
A:將120g球形石墨超聲分散於100ml無水乙醇中,然後將矽粉15g加入石墨分散液中,採用行星式球磨機球磨7h,球磨機轉速為250r/min,然後放置真空乾燥箱中80℃真空乾燥3h;納米矽在進行球磨前,先將矽粉添加至其12倍質量的濃度為12wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至130℃,保溫疏解8h;疏解後過濾得到的矽粉添加至其12倍質量的濃度為80wt%的乙醇溶液中,加熱至90℃後保溫5h,再次進行過濾,打漿至漿料叩解度為16°SR,然後進行磨漿,磨漿至漿料叩解度為22°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其22倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.6倍的海藻提取液,海藻提取液的固含量為35wt%,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為4wt%的氯化鈣溶液,在50℃下交聯2h後,轉移液氮中冰凍2天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽;
B:將20g中溫煤瀝青與上述A中製備的矽/石墨複合物於錐形混合機中進行混合,混合時間為30min;然後將上述混合物置於臥式機械融合機內進行機械融合,機械融合機的轉速為800r/min,融合時間為25min;最後將融合後的混合物轉移至管式爐中高溫炭化處理,溫度程序為:室溫下以2℃/min的速率升溫至500℃,保溫30min,然後以5℃/min的速率升溫至1000℃,保溫180min,最後自然冷卻至室溫,粉碎過篩即可得到矽碳複合材料。
如實施例1中所述進行電極片的製作,組裝成扣式電池,首次循環採用0.05C倍率進行活化,之後採用0.2C倍率充放電電壓範圍為0.01-1.5V進行充放電循環測試,測試結果見表1。
實施例3:
一種矽碳複合負極材料的製備方法,包括以下步驟:
A:將80g球形石墨超聲分散於100ml無水乙醇中,然後將矽粉10g加入石墨分散液中,採用行星式球磨機球磨3h,球磨機轉速為250r/min,然後放置真空乾燥箱中80℃真空乾燥3h;納米矽在進行球磨前,先將矽粉添加至其12倍質量的濃度為12wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至130℃,保溫疏解8h;疏解後過濾得到的矽粉添加至其12倍質量的濃度為80wt%的乙醇溶液中,加熱至90℃後保溫5h,再次進行過濾,打漿至漿料叩解度為16°SR,然後進行磨漿,磨漿至漿料叩解度為22°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其22倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.6倍的海藻提取液,海藻提取液的固含量為35wt%,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為4wt%的氯化鈣溶液,在50℃下交聯2h後,轉移液氮中冰凍2天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽;
B:將10g中溫煤瀝青與上述A中製備的矽/石墨複合物於錐形混合機中進行混合,混合時間為30min;然後將上述混合物置於臥式機械融合機內進行機械融合,機械融合機的轉速為500r/min,融合時間為15min;最後將融合後的混合物轉移至管式爐中高溫炭化處理,溫度程序為:室溫下以0.5℃/min的速率升溫至500℃,保溫1min,然後以0.5℃/min的速率升溫至1000℃,保溫10min,最後自然冷卻至室溫,粉碎過篩即可得到矽碳複合材料。
如實施例1中所述進行電極片的製作,組裝成扣式電池,首次循環採用0.05C倍率進行活化,之後採用0.2C倍率充放電電壓範圍為0.01-1.5V進行充放電循環測試,測試結果見表1。
實施例4:
一種矽碳複合負極材料的製備方法,包括以下步驟:
A:將30g鱗片石墨超聲分散於100ml無水乙醇中,然後將矽粉40g加入石墨分散液中,採用行星式球磨機球磨10h,球磨機轉速為250r/min,然後放置真空乾燥箱中80℃真空乾燥3h;納米矽在進行球磨前,先將矽粉添加至其8倍質量的濃度為8wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至110℃,保溫疏解6h;疏解後過濾得到的矽粉添加至其8倍質量的濃度為70wt%的乙醇溶液中,加熱至80℃後保溫3h,再次進行過濾,打漿至漿料叩解度為14°SR,然後進行磨漿,磨漿至漿料叩解度為18°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其18倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.4倍的海藻提取液,海藻提取液的固含量為25wt%,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為2wt%的氯化鈣溶液,在40℃下交聯1h後,轉移液氮中冰凍1天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽。
B:將30g中溫煤瀝青與上述A中製備的矽/石墨複合物於錐形混合機中進行混合,混合時間為30min;然後將上述混合物置於臥式機械融合機內進行機械融合,機械融合機的轉速為1000r/min,融合時間為60min;最後將融合後的混合物轉移至管式爐中高溫炭化處理,溫度程序為:室溫下以5℃/min的速率升溫至500℃,保溫200min,然後以10℃/min的速率升溫至1200℃,保溫240min,最後自然冷卻至室溫,粉碎過篩即可得到矽碳複合材料。
如實施例1中所述進行電極片的製作,組裝成扣式電池,首次循環採用0.05C倍率進行活化,之後採用0.2C倍率充放電電壓範圍為0.01-1.5V進行充放電循環測試,測試結果見圖2與表1。
實施例5
一種矽碳複合負極材料的製備方法,包括以下步驟:
A:將120g球形石墨超聲分散於100ml無水乙醇中,然後將矽粉40g加入石墨分散液中,採用行星式球磨機球磨7h,球磨機轉速為250r/min,然後放置真空乾燥箱中80℃真空乾燥3h;納米矽在進行球磨前,先將矽粉添加至其8倍質量的濃度為8wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至110℃,保溫疏解6h;疏解後過濾得到的矽粉添加至其8倍質量的濃度為70wt%的乙醇溶液中,加熱至80℃後保溫3h,再次進行過濾,打漿至漿料叩解度為14°SR,然後進行磨漿,磨漿至漿料叩解度為18°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其18倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.4倍的海藻提取液,海藻提取液的固含量為25wt%,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為2wt%的氯化鈣溶液,在40℃下交聯1h後,轉移液氮中冰凍1天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽。
B:將55g高溫煤瀝青與上述A中製備的矽/石墨複合物於錐形混合機中進行混合,混合時間為30min;然後將上述混合物置於臥式機械融合機內進行機械融合,機械融合機的轉速為800r/min,融合時間為25min;最後將融合後的混合物轉移至管式爐中高溫炭化處理,溫度程序為:室溫下以2℃/min的速率升溫至500℃,保溫30min,然後以5℃/min的速率升溫至1000℃,保溫180min,最後自然冷卻至室溫,粉碎過篩即可得到矽碳複合材料。
如實施例1中所述進行電極片的製作,組裝成扣式電池,首次循環採用0.05C倍率進行活化,之後採用0.2C倍率充放電電壓範圍為0.01-1.5V進行充放電循環測試,測試結果見表1。
實施例6
一種矽碳複合負極材料的製備方法,包括以下步驟:
A:將120g球形石墨超聲分散於100ml無水乙醇中,然後將矽粉40g加入石墨分散液中,採用行星式球磨機球磨7h,球磨機轉速為250r/min,然後放置真空乾燥箱中80℃真空乾燥3h;納米矽在進行球磨前,先將矽粉添加至其10倍質量的濃度為10wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至120℃,保溫疏解7h;疏解後過濾得到的矽粉添加至其10倍質量的濃度為75wt%的乙醇溶液中,加熱至85℃後保溫4h,再次進行過濾,打漿至漿料叩解度為15°SR,然後進行磨漿,磨漿至漿料叩解度為20°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其20倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.5倍的海藻提取液,海藻提取液的固含量為30wt%,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為3wt%的氯化鈣溶液,在45℃下交聯1.5h後,轉移液氮中冰凍2天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽。
B:將58g焦油瀝青與上述A中製備的矽/石墨複合物於錐形混合機中進行混合,混合時間為30min;然後將上述混合物置於臥式機械融合機內進行機械融合,機械融合機的轉速為800r/min,融合時間為25min;最後將融合後的混合物轉移至管式爐中高溫炭化處理,溫度程序為:室溫下以2℃/min的速率升溫至500℃,保溫30min,然後以5℃/min的速率升溫至1000℃,保溫180min,最後自然冷卻至室溫,粉碎過篩即可得到矽碳複合材料。
如實施例1中所述進行電極片的製作,組裝成扣式電池,首次循環採用0.05C倍率進行活化,之後採用0.2C倍率充放電電壓範圍為0.01-1.5V進行充放電循環測試,測試結果見表1。
實施例7
一種矽碳複合負極材料的製備方法,包括以下步驟:
A:將120g球形石墨超聲分散於100ml無水乙醇中,然後將納米矽40g加入石墨分散液中,採用行星式球磨機球磨7h,球磨機轉速為250r/min,然後放置真空乾燥箱中80℃真空乾燥3h;
納米矽在進行球磨前,先將矽粉添加至其12倍質量的濃度為12wt%的氫氧化鈉溶液中,分散均勻後,減壓加熱至130℃,保溫疏解8h;疏解後過濾得到的矽粉添加至其12倍質量的濃度為80wt%的乙醇溶液中,加熱至90℃後保溫5h,再次進行過濾,打漿至漿料叩解度為16°SR,然後進行磨漿,磨漿至漿料叩解度為22°SR,打漿後用水洗淨並減壓蒸餾後得到的矽粉添加至其22倍質量的丁二腈中,得到懸浮液;向懸浮液中添加其體積0.6倍的海藻提取液,海藻提取液的固含量為35wt%,攪拌均勻得到混合溶液,然後向混合溶液中添加等體積的濃度為4wt%的氯化鈣溶液,在50℃下交聯2h後,轉移液氮中冰凍2天,然後取出冰凍物,用流水解凍,製得納米矽凝膠,再用無水乙醇進行溶劑置換,最後經過真空乾燥和研磨後,製得納米矽。
B:將60g中溫煤瀝青與上述A中製備的矽/石墨複合物於錐形混合機中進行混合,混合時間為30min;然後將上述混合物置於臥式機械融合機內進行機械融合,機械融合機的轉速為800r/min,融合時間為25min;最後將融合後的混合物轉移至管式爐中高溫炭化處理,溫度程序為:室溫下以2℃/min的速率升溫至500℃,保溫30min,然後以5℃/min的速率升溫至1000℃,保溫180min,最後自然冷卻至室溫,粉碎過篩即可得到矽碳複合材料。
為了檢驗本發明鋰離子電池矽碳複合負極材料的性能,組裝成半電池對其進行性能測試。將所得的鋰離子電池矽碳複合負極材料分別與導電劑碳納米管、粘結劑CMC和SBR按照質量比80:10:10球磨混合,用去離子水調節混合物的粘度製成漿料,均勻塗覆在銅箔上,
80℃真空乾燥8h,製得實驗電池用極片。再以鋰片作為對電極在手套箱中組裝成CR2032型扣式電池,首個循環採用0.05C倍率下進行活化,之後採用0.2C倍率充放電電壓範圍為0.01-1.5V進行循環性能測試,測試結果見表1。
實施例8
其他條件與實施例1相同,不同之處在於組裝電池時把粘結劑CMC+SBR改為海藻酸鈉,測試結果見圖3與表1。
實施例9
其他條件與實施例1相同,不同之處在於組裝電池時把粘結劑CMC改為PVDF,且溶劑由去離子水改為N-甲基吡咯烷酮,測試結果見表1。
實施例1-9的相關性能數據列於下表1中。
表1、性能數據
從表1中可以看出,本發明所述方法製備的矽碳複合負極材料具有優異的電化學性能,首次庫侖效率高、壓實密度高、循環性能穩定。
本發明通過上述實施例和對比例來描述本發明的詳細工藝流程,但本發明並不限於上述詳細工藝流程,上述的具體實施方式僅僅是示意性的,而不是限制性的,所屬技術領域的技術人員應該明白,對本發明的任何改進,對本發明產品各原料的等效替換及輔助成分的添加、具體方式的選擇等,均落在本發明的保護範圍和公開範圍之內。