一種四甲基羅丹明的製備工藝的製作方法
2023-11-10 01:37:17 3
本發明涉及一種螢光染料的製備工藝,具體涉及一種四甲基羅丹明的製備工藝。
背景技術:
羅丹明類化合物是以氧雜蒽為母體的染料,由於其特殊的結構,羅丹明類螢光染料與其它常用的螢光染料相比,具有如下優點:光穩定性好、對ph不敏感、較寬的波長範圍和較高的螢光量子產率。因此被廣泛應用在藥理學、生理學、分子生物學、細胞生物學、分子遺傳學、環境化學、單個分子檢測、信息科學、螢光標記、雷射染料等方面。其中,四甲基羅丹明(tamra)有親脂性強、斯託克斯位移(stokesshift)較小、螢光量子產率較高、發射波長較長以及吸光係數較大的優點,被廣泛應用於dna測序、免疫螢光染色和標記抗體等方面(u.s.patent,637290781)。此外,四甲基羅丹明(tamra)還可以被廣泛應用於研究蛋白質的結構、性能、內部的相互作用關係,其用來分析研究粘度比較大、分子量大於4×106的蛋白質效果很好,並且四甲基羅丹明分析速度快且不會汙染蛋白多糖結構,而其它類探針很難對該蛋白進行分析檢測;因此,四甲基羅丹明是分析化學和生物醫藥科學等生物技術領域中最常用的螢光染料(anal.biolchem.;1995,227,394~396)。
四甲基羅丹明有著十分廣闊的應用前景,現有的製備方法包括傳統的直接縮合合成法、分步縮合合成法、氨基酚與醛基化合物的縮合、氨基酚與1-羥基-3氧代-1,3-二氫異苯並呋喃-5-羧酸縮合氧化反應;上述製備方法收率較低(一般只有百分之十幾),純化過程繁瑣(製備柱純化),致使四甲基羅丹明的價格十分昂貴。
其中傳統的直接縮合合成法是以n,n-二甲基間胺基苯酚和偏苯酸三酐為原料,在布朗斯德酸或者路易斯酸中高溫縮合製得,(thejournaloforganicchemistry,1989,54(23):5642-5644;j.chem.soc,perkintransii,1993:1195;curr.org.chem.2016,20,1584.)。該方法的優點是原料易得,都是工業化產品,成本低廉;但四甲基羅丹明的收率較低,一般只有百分之十幾;並且對反應條件的要求比較苛刻,一般都需要高溫甚至熔融。該合成法首先得到的是5位取代基和6位取代基的異構體混合物,這類5位和6位取代基異構體大多採用色譜方法進行分離(如製備柱或者矽膠柱層析),該分離方法除成本高,並且只能得到克級甚至毫克級的成品,難於批量製備。
技術實現要素:
基於上述背景技術,本發明的目的在於一種原子經濟性高,操作簡便,低成本,易於大規模生產的四甲基羅丹明(tamra)製備工藝;本發明以n,n-二甲基間胺基苯酚和偏苯酸三酐為原料,依次進行環合反應處理,純化處理,拆分反應處理及酸析反應處理,即得到四甲基羅丹明。
即本發明是通過環合反應直接製得到5位取代基和6位取代基的5(6)-四甲基羅丹明異構體混合物,混合物與有機胺反應形成成鹽,通過這種鹽在溶劑中的溶解性不同,從而達到分離純化的目的。
所述的一種四甲基羅丹明(tamra)製備工藝,具體包括如下步驟:
1)5(6)-四甲基羅丹明的合成:取n,n-二甲基間胺基苯酚、偏苯酸三酐及第一溶劑混合,加入酸反應,即得5(6)-四甲基羅丹明;
2)5(6)-四甲基羅丹明銨鹽製備:將步驟1)得到的5(6)-四甲基羅丹明加入第二溶劑中溶解,加入有機胺反應,過濾,濾餅烘乾即得5-四甲基羅丹明銨鹽,濾液濃縮即得6-四甲基羅丹明銨鹽;
3)5-四甲基羅丹明和6-四甲基羅丹明的製備:以步驟2)得到的5-四甲基羅丹明銨鹽和6-四甲基羅丹明銨鹽分別與第三溶劑混合,得到的溶液分別加入酸反應,過濾,濾餅烘乾即得5-四甲基羅丹明和6-四甲基羅丹明。
優選地,所述製備工藝包括以下步驟:
1)5(6)-四甲基羅丹明的合成:取n,n-二甲基間胺基苯酚、偏苯酸三酐及第一溶劑混合,加入酸,在120~220℃下,反應2~8小時,過濾,即得5(6)-四甲基羅丹明(5,6-isomer,異構體混合物);
2)5(6)-四甲基羅丹明銨鹽製備:將步驟1)得到的5(6)-四甲基羅丹明加入第二溶劑溶解,加入有機胺,在25~40℃下,反應0.5~4小時,過濾,濾餅烘乾即得5-四甲基羅丹明銨鹽,濾液濃縮即得6-四甲基羅丹明銨鹽;
3)5-四甲基羅丹明和6-四甲基羅丹明的製備:以步驟2)得到的5-四甲基羅丹明銨鹽和6-四甲基羅丹明銨鹽分別與第三溶劑混合,得到的溶液分別加入酸,在0~25℃下,反應0.5~3小時,過濾,濾餅烘乾即得5-四甲基羅丹明和6-四甲基羅丹明;
進一步優選地,所述製備工藝包括以下步驟:
1)5(6)-四甲基羅丹明的合成:取n,n-二甲基間胺基苯酚、偏苯酸三酐及第一溶劑混合,加入酸,在140~160℃下,反應3~5小時,過濾,即得5(6)-四甲基羅丹明;
2)5(6)-四甲基羅丹明銨鹽製備:將步驟1)得到的5(6)-四甲基羅丹明加入第二溶劑溶解,加入有機胺,在25~30℃下,反應1~3小時,過濾,濾餅烘乾即得5-四甲基羅丹明銨鹽,濾液濃縮即得6-四甲基羅丹明銨鹽;
3)5-四甲基羅丹明和6-四甲基羅丹明的製備:以步驟1)得到的5-四甲基羅丹明銨鹽和6-四甲基羅丹明銨鹽分別與第三溶劑混合,得到的溶液分別加入酸,在0~25℃下,反應1.5~2.5小時,過濾,濾餅烘乾即得5-四甲基羅丹明和6-四甲基羅丹明。
所述n,n-二甲基胺基苯酚與偏苯三酸酐的質量比為2.05~2.5:1。
步驟1)中所述第一溶劑選自乙腈、甲苯、二甲苯、氯苯或鄰二氯苯中的一種,優選為鄰二氯苯;
步驟1)中所述第一溶劑與偏苯三酸酐的質量比為1~20:1,優選為10~12:1。
步驟1)中所述酸選自甲磺酸、三氟甲磺酸,硫酸或磷酸中的一種,優選為甲磺酸或三氟甲磺酸
步驟1)中所述酸與偏苯三酸酐的質量比為1~10:10;優選為2~5:10。
步驟2)中所述5(6)-四甲基羅丹明、所述有機胺、所述第二溶劑的質量比為1∶2~5∶2~10;
所述有機胺選自二甲胺,二乙胺,三乙胺,正丁胺,二異丙胺或二異丙基乙基胺中的一種,優選為二異丙胺;
步驟2)中所述第二溶劑選自乙腈、四氫呋喃、二氧六環、n,n-二甲基甲醯胺或乙二醇二甲醚中的一種,優選為二氧六環。
步驟3)中5-四甲基羅丹明二異丙基胺鹽或6-四甲基羅丹明二異丙基胺鹽與所酸、所述第三溶劑的質量比互相獨立地為1:2~5:2~10;所述酸的濃度為2~5mol/l;
步驟3)中所述酸選自鹽酸,硫酸,硝酸,磷酸,甲磺酸或三氟乙酸中的一種,優選為鹽酸。
步驟3)中所述第三溶劑選自甲醇、乙醇、異丙醇、乙二醇或水中的一種,優選為水。
進一步地,本發明提出的一種四甲基羅丹明的製備工藝,其中,所述5(6)-四甲基羅丹明的合成後還包括5(6)四甲基羅丹明的純化處理:
所述純化處理具體為:將步驟1)製得的5,6-四甲基羅丹與純化溶劑混合,攪拌,靜置,過濾即得5(6)-四甲基羅丹明純品;
優選地,所述5(6)-四甲基羅丹明的純化具體為:將步驟1)製得的5(6)-四甲基羅丹與純化溶劑混合,在40~80℃下,攪拌45~90分鐘,室溫靜置12~24小時,過濾得到5,6-四甲基羅丹明純品;
進一步優選地,所述5(6)-四甲基羅丹明的純化具體為:將步驟1)製得的5(6)-四甲基羅丹與純化溶劑混合,在70~80℃下,攪拌50~70分鐘,室溫靜置12~14小時,過濾得到5,6-四甲基羅丹明純品。
所述純化溶劑選自乙腈、四氫呋喃、二氧六環、乙醇、或乙二醇二甲醚中的一種,優選為乙腈;
所述純化溶劑與5(6)-四甲基羅丹明的質量比為1~10:1,優選為3~6:1。
進一步地,本發明提出的一種四甲基羅丹明的製備工藝,其中,所述5(6)-四甲基羅丹明的合成及5(6)-四甲基羅丹明的純化在氮氣氣氛中進行。
作為優選方案,本發明提供的一種四甲基羅丹明的製備工藝包括以下步驟:
1)5(6)-四甲基羅丹明的合成:取n,n-二甲基間胺基苯酚、偏苯酸三酐及二氯苯混合,在氮氣氣氛下,加入甲磺酸,在140~160℃下,反應3~5小時,過濾,即得5(6)-四甲基羅丹明粗品;
2)5(6)-四甲基羅丹明的純化:將步驟1)製得的5(6)-四甲基羅丹粗品與乙腈混合,在氮氣氣氛下,加熱至70~80℃,攪拌50~70分鐘,室溫靜置12~14小時,過濾得到5(6)-四甲基羅丹明純品;
3)5(6)-四甲基羅丹明銨鹽製備:將步驟2)得到的5(6)-四甲基羅丹明純品加入二氧六環中溶解,加入二異丙胺,在25~30℃下,反應1~3小時,過濾,濾餅烘乾即得5-四甲基羅丹明銨鹽,濾液濃縮即得6-四甲基羅丹明銨鹽;
4)5-四甲基羅丹明和6-四甲基羅丹明的製備:以步驟3)得到的5-四甲基羅丹明銨鹽和6-四甲基羅丹明銨鹽分別與水混合,在0~25℃下,反應1.5~2.5小時,過濾,濾餅烘乾即得5-四甲基羅丹明和6-四甲基羅丹明。
本發明的至少具有如下有益結果:
1)本發明採用的是n,n-二甲基間胺基苯酚和偏苯酸三酐為原料,直接環合法合成中間體5(6)-四甲基羅丹明,與一般環合法不同的是採用的高沸點溶劑作為環合媒介,而且環合產物在溶劑中的溶解度較差,從而在反應體系中沉澱出來,從而顯著的提高反應轉化率和提純的簡便性。
2)本發明採用重結晶的方法,將步驟1)製得的5(6)-四甲基羅丹明粗品,能夠高效的提純。
3)本發明通過5(6)-四甲基羅丹明與有機胺成鹽,並利用異構體銨鹽在溶劑中溶解性的不同,從而達到分離兩個異構體的目的,極大降低了分離成本,簡化了操作步驟,從而使四甲基羅丹明工業化生產成為可能。
4)本發明採用酸析的方法,將製得的5-四甲基羅丹明銨鹽和6-四甲基羅丹明銨鹽高效轉化為5-四甲基羅丹明和6-四甲基羅丹明。
5)本發明操作簡便、對設備要求低、成本低,設備使用效率高等特點,達到工業化生產應用的要求。
附圖說明
圖1為5(6)-四甲基羅丹明核磁氫譜圖
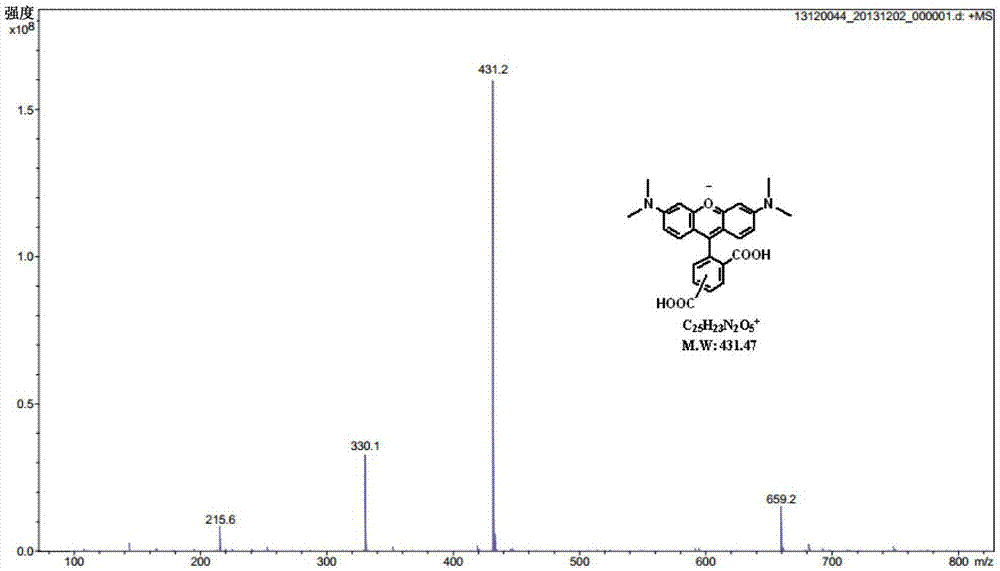
圖2為5(6)-四甲基羅丹明質譜圖
圖3為5-四甲基羅丹明核磁氫譜圖
圖4為5-四甲基羅丹明質譜圖
圖5為6-四甲基羅丹明核磁氫譜圖
圖6為6-四甲基羅丹明質譜圖
具體實施方式
以下實施例用於說明本發明,但不用來限制本發明的範圍。
實施例所用試劑未加特別說明均為市售分析純,使用前未經特別純化。
實施例1
5(6)-四甲基羅丹明的合成:將4千克n,n-二甲基間胺基苯酚和2680克偏苯酸三酐,以及20l鄰二氯苯加入50l帶有溫度計和攪拌的玻璃反應釜內。抽去反應釜中的空氣,通入氮氣,反應體系逐漸升溫至40℃,加入500克甲磺酸,反應體系升溫至150℃,反應4h,觀察發現有大量的固體生成,從體系中沉澱出來並黏附在反應釜內壁上。停止反應後,靜置15min,反應完成。
取樣檢測,液相相色譜結果表明,偏苯酸三酐的轉化率為99%,5(6)-四甲基羅丹明收率為82%。由反應釜下層放料口將放出鄰二氯苯溶劑,加入20l甲醇至反應釜內,攪拌並緩慢加熱至50℃,帶溶劑將黏附在反應釜內壁上固體溶解完全,完全溶解大約需2小時,將甲醇溶液放出並濃縮,即得到4780克5(6)-四甲基羅丹明粗品。
實施例2
5(6)-四甲基羅丹明的純化:將實施例1所得的4780克5(6)-四甲基羅丹明粗品與20l乙腈加入50l帶有溫度計和攪拌的玻璃反應釜中。抽去反應釜中的空氣,通入氮氣,反應體系逐漸升溫至80℃,熱熔約1小時,體系中的固體溶解完全,之後停止加熱,停止攪拌,靜置至室溫,發現有固體析出,繼續靜置12小時,過濾;濾液濃縮後再重複這一過程,合併兩次的濾餅,用冷卻的2l乙腈洗滌兩次,烘乾得4386克5(6)-四甲基羅丹明純品,為墨綠色固體,收率72%,純度97.6%,其中5-四甲基羅丹佔比54.49%,6-四甲基羅丹佔比43.04%。
如圖1和圖2所示,1h-nmr:(400mhz,cd3od,ppm)δ:6.983-6.988(d,4h,j=9.3),7.052-7.077(m,4h,j=11.2),7.129-7.137(m,4h,j=12.3),7.535-7.555(d,2h,j=5.9),7.978-7.982(d,1h,j=3.7),8.411-8.431(m,1h,j=7.8),8.920-8.923(d,1h,j=3.5).esi-ms(m/z):431,m+。
實施例3
5,6-四甲基羅丹明銨鹽製備:將實施例2製得的4300克5(6)-四甲基羅丹明純品與43l二氧六環加入100l帶有溫度計和攪拌的玻璃反應釜內,加熱至40℃攪拌2小時,使固體充分溶解,待固體充分溶解後,停止加熱,反應體系逐漸冷卻至室溫,滴加加入3036克二異丙胺,滴加過程,放熱,控制反應體系溫度不超過30℃。體系逐漸變為紫黑色溶液,滴加完畢繼續攪拌1小時,體系變為紫紅色懸浮液,停止攪拌,靜置1小時,發現有大量紅色固體析出,繼續靜置2小時,抽濾,其中濾餅為5-四甲基羅丹明二異丙胺鹽,濾餅用5l二氧六環洗滌後,在60℃下乾燥5h,得到5-四甲基羅丹明二異丙胺鹽2434g。
合併濾液,其中濾液中5-四甲基羅丹明二異丙胺鹽佔15.71%,6-四甲基羅丹明二異丙胺鹽佔80.77%;濃縮旋幹濾液,重複以上操作一次,得到5-四甲基羅丹明二異丙胺鹽115g,合併得到2549克,收率88%,hplc=97.8%。母液濃縮旋幹後用25l乙腈打漿得到6-四甲基羅丹明二異丙胺鹽2080克,收率91%,hplc=98.6%.
如圖3和圖4所示,1h-nmr:(400mhz,cd3od,ppm)δ:6.985(s,2h),7.072-7.078(dd,2h,j=8.7),7.130-7.153(dd,2h,j=10.3),7.534-7.554(d,1h,j=5.7),8.425-8.449(d,1h,j=7.8),8.919(s,1h).esi-ms(m/z):431,m+。
hplc97.37%,收率為:86%。
實施例4
5-四甲基羅丹明和6-四甲基羅丹明的製備:將實施例3製得的2549克5-四甲基羅丹明二異丙胺鹽,8l水加入25l帶有溫度計和攪拌的玻璃反應釜,25℃攪拌1小時,使固體充分溶解,待固體充分溶解後,滴加加入4l3mol/l鹽酸,有紅色固體析出,滴加完畢繼續攪拌1小時,抽濾,得到粘稠狀褐紅色固體5-四甲基羅丹明1775克。
將實施例3製得的2080克6-四甲基羅丹明二異丙胺鹽與7l水加入25l帶有溫度計和攪拌的玻璃反應釜,25℃攪拌1小時,使固體充分溶解,待固體充分溶解後,滴加加入3.l3mol/l的鹽酸,有紅色固體析出,滴加完畢繼續攪拌1小時,抽濾,得到粘稠狀褐紅色固體6-四甲基羅丹明1465克。
如圖5和圖6所示,1h-nmr:(400mhz,cd3od,ppm)δ:6.972(s,2h),7.112-7.148(d,2h,j=8.3),7.160-7.178(d,2h,j=8.7),7.975(s,1h),8.404-8.412(d,2h,j=5.3).esi-ms(m/z):431,m+。
hplc98.89%,收率為:87%。
雖然,上文中已經用一般性說明、具體實施方式及試驗,對本發明作了詳盡的描述,但在本發明基礎上,可以對之作一些修改或改進,這對本領域技術人員而言是顯而易見的。因此,在不偏離本發明精神的基礎上所做的這些修改或改進,均屬於本發明要求保護的範圍。