氮化矽襯底的製造方法與流程
2023-12-03 02:09:01 1
本發明涉及一種氮化矽襯底的製造方法。
背景技術:
近些年,隨著電子設備、半導體裝置的高集成化、高電力化,從半導體元件產生的熱的散熱技術正變得極為重要。因此,對於在電子設備或半導體裝置中被用作絕緣部件的襯底等,尋求散熱性優異的散熱襯底。
作為散熱襯底的材料例如考慮金屬或陶瓷等,但金屬的耐氧化性、耐水性、耐腐蝕性不如陶瓷,特別是在超過50℃的條件下不可能不進行冷卻來使用。另外,由於具有導電性,因此難以用作要求絕緣的高密度裝配襯底等要求較高散熱性的絕緣襯底。
另一方面,與金屬相比,陶瓷具有較高的耐氧化性、耐水性、耐腐蝕性,例如氧化鋁、氮化鋁等被用作散熱襯底的材料。其中氮化鋁同時具有優異的絕緣性和高熱導率,因此被用作功率模塊的散熱襯底材料。然而,由於氮化鋁的強度、斷裂任性等機械特性較差、缺乏可靠性,因此其用途非常有限。
另一方面,氮化矽燒結體作為同時具有高強度和高韌性的優異構造用陶瓷材料被廣為人知,其預測其單晶的熱導率具有200~320W/mK的極為高的值。因此,期待將其用作散熱襯底的材料。然而,對於一般的氮化矽燒結體,在氮化矽顆粒內部固溶有雜質氧等。因此,用於導熱的聲子會被固溶,與單晶時所預測的20~80W/mK的值相比,氮化矽燒結體的熱導率會低很多。
在此,在非專利文獻1公開了為了在氮化矽燒結體中實現較高的熱導率,尋求在燒結時降低低熱導玻璃相、以及減少在氮化矽顆粒內部固溶的氧。
然而,氮化矽是共價性極其高的難燒結性材料。因此,為了得到緻密的燒結體,需要進行使用燒結助劑的液相燒結。
並且,作為在製造氮化矽的燒結體時所添加的燒結助劑可舉出氧化物。所添加的燒結助劑通過在燒結中與存在於氮化矽粉末表面的矽石反應而生成液相,利用此液相來進行緻密化和晶粒生長。對於在燒結中生成的液相,在冷卻時其大部分作為玻璃相殘留在燒結體中。
另外,在製造氮化矽的燒結體時,作為用於氮化矽燒結體的高導熱性而添加的燒結助劑,可舉出氧親和力較高的稀土氧化物。當添加了稀土氧化物時,由於具有所生成的液相捕捉大量的氧的效果,因此能夠降低氮化矽顆粒內部的固溶氧量,能夠實現高導熱性。
然而,當作為燒結助劑僅添加稀土氧化物時,由於所生成的液相的熔點較高,因此難以得到機械特性優異的緻密的燒結體。因此,為了得到使機械特性和高熱導率同時優異的氮化矽燒結體,採取了各種方法。
例如,在專利文獻1中公開了一種高熱導率氮化矽燒結體的製造方法,其在向Al含量為0.1重量%以下的氮化矽粉末中添加1重量%以上15重量%以下的選自Mg、Ca、Sr、Ba、Y、La、Ce、Pr、Nd、Sm、Gd、Dy、Ho、Er、Yb中的一種或兩種以上的元素的氧化物燒結助劑並成形後,在1氣壓以上500氣壓以下的氮氣壓下,以1700℃以上2300℃以下的溫度,燒結直至孔隙率為5%以下且發現預定的組織。並且,其公開了得到了熱導率為80W/(m K)以上、斷裂韌性為7MPam1/2以上、以四點彎曲法測定的彎曲強度為600MPa以上的高熱導率氮化矽燒結體。
另外,在專利文獻中公開了一種高熱導氮化矽燒結體的製造方法,其對在氮化矽粉末中添加了總計1.0重量%以下的鎂及釔及/或鑭族元素的一種以上的氧化物的原料粉末進行成形後,在溫度1800℃~2000℃、氮氣壓0.5MPa~10Mpa下,使用由氮化矽、氮化硼及氧化鎂構成的混合粉末燒結成燒結氣氛調節用的填裝粉。另外,公開了通過將燒結體的顆粒的大小、氮化矽顆粒內的氧的量、殘留燒結助劑成分量設為預定的範圍,從而得到常溫下熱導率為90W/mK以上、3點彎曲強度為600MPa以上的氮化矽燒結體。
另外,在非專利文獻2中,公開了在以往的燒結法中,儘管燒結時間的增加、氮化矽的晶粒生長被促進、熱導率提高,但由於過度的晶粒生長,使得熱導率提高的同時,強度及斷裂韌性顯著的降低。
如上述事例所列舉的那樣,在以往的方法中,通過使用氮化矽粉末、使燒結助劑的種類、添加量、燒結條件等工藝參數最佳化、實現預定的細微構造,從而製作了具有80W/mK以上的熱導率以及600MPa以上的彎曲強度的氮化矽燒結體。
然而,即使是在專利文獻1、2所公開的任意的製造方法中,由於使用昂貴的氮化矽粉末,因此存在製造成本升高的問題。另外,當改變燒結條件以使熱導率提高到超過100W/mK時,存在強度、韌性急劇降低、機械性的可靠性缺乏的問題。
這樣一來,從同時滿足製造成本和機械特性的兩個觀點來看,利用以往的燒結法得到的高熱導氮化矽並未滿足該要求。
因此,從降低原料粉末所需的成本的觀點來看,正在進行一種高熱導氮化矽材料的開發,該高熱導氮化矽材料使用便宜的矽粉末作為原料粉末,在氮中對其成形體進行氮化後,在高溫下進行燒結、也即使用了反應燒結手法。
例如專利文獻3中公開了一種Si3N4燒結體的製造方法,其將含氧量為1重量%以下的80~99重量%的Si粉末、和1~20重量%的Y、Yb、Sm的至少一種元素的氧化物粉末混合,在氮氣氛中以1400℃以下的溫度對其成型體進行氮化處理後,在含氮氣氛中以1700℃~1950℃的溫度對所得到的氮化體進行燒結。在該Si3N4燒結體的製造方法中,通過使用含氧量為1重量%以下的高純度的Si粉末,從而抑制雜質氧離子向Si3N4結晶顆粒內的固溶、並實現Si3N4燒結體的高導熱化。另外,還記載了以下內容,通過在原料粉末中進一步添加混合Si粉末的1~10重量%的還原性塗劑,在100Torr以下的真空中或含氮氣氛中以200℃~800℃的溫度對其成形體進行熱處理後,進行上述氮化處理及燒結,從而進一步減少所得到Si3N4燒結體中的含氧量,並進一步提高熱導率。
另外,專利文獻4和5中公開了在矽粉末或矽粉末與氮化矽粉末的混合粉末中以將矽換算成氮化矽時的比例混合0.5~7摩爾%的稀土元素的氧化物和1~7摩爾%的鎂化合物,以及通過將該混合物成形並氮化、在預定壓力的氮中對所得到的氮化體進行加熱並以相對密度為95%以上的方式進行緻密化從而製造氮化矽燒結體。並且公開了得到具有熱導率為100W/mK以上、三點彎曲強度為600MPa以上、斷裂韌性值為7MPam1/2以上的特性的氮化矽燒結體。
然而,根據專利文獻3~5公開的氮化矽燒結體的製造方法製造氮化矽襯底時,在氮化矽襯底的表層部以90μm~140μm左右的厚度產生了包含多個氣孔的多孔質的改性層。當在氮化矽襯底的表面包含改性層時,由於該氮化矽襯底的電氣特性及機械強度降低,因此需要通過研磨等將形成在氮化矽襯底表面上的改性層部分刮掉,存在製造工序增加、以及隨之而來的成本增大的問題。
專利文獻1:(日本)特開平9-30866號公報
專利文獻2:(日本)特開2002-293642號公報
專利文獻3:(日本)特開平11-314969號公報
專利文獻4:(日本)專利第5046221號
專利文獻5:(日本)專利第4997431號
非專利文獻1:Journal of the American Ceramic Society,「Thermal Conductivity of be-ta-Si3N4II:Effect of Lattice Oxygen,」83[8]1985-1992(2000)
非專利文獻2:Y.Hayashi et al.,J.Ceram.Soc.Japan,109,p.1046(2001)
技術實現要素:
鑑於上述現有技術的問題,本發明的目的在於提供一種氮化矽襯底的製造方法,其能夠從包含矽粉末的原料粉末來進行製造,並能夠製造在燒結體形成後無需除去改性層的緻密的氮化矽襯底。
本發明提供一種氮化矽襯底的製造方法,其包括:原料粉末準備步驟,其準備含有矽粉末、稀土元素化合物、及鎂化合物的原料粉末,其中,當將原料粉末中的矽換算成氮化矽時,該原料粉末含有以氧化物計1摩爾%以上7摩爾%以下的所述稀土元素化合物,並含有以氧化物計8摩爾%以上15摩爾%以下的所述鎂化合物;片材成形步驟,其將所述原料粉末成形為片狀而形成片材體;氮化步驟,其在氮氣氛中以1200℃以上1500℃以下對所述片材體進行加熱,將包含在片材體中的矽氮化;以及燒結步驟,其在氮氣氛下對完成了所述氮化步驟的所述片材體進行燒結。
根據本發明,能夠提供一種氮化矽襯底的製造方法,其能夠從包含矽粉末的原料粉末來進行製造,並能夠製造在燒結體形成後無需除去改性層的緻密的氮化矽襯底。
附圖說明
圖1是本發明的實施例1中所得到的氮化矽襯底剖面的SEM圖像。
圖2A是比較例1中所得到的氮化矽襯底剖面的SEM圖像。
圖2B是比較例1中所得到的氮化矽襯底剖面的SEM圖像。
圖3是比較例2中所得到的氮化矽襯底剖面的SEM圖像。
具體實施方式
以下,對本發明的實施方式進行說明,本發明不限定於以下實施方式,可以在不脫離本發明的範圍的情況下,對以下實施方式進行各種變形及置換。
在本實施方式中,對本發明的氮化矽襯底的製造方法的一個結構例進行說明。
本實施方式的氮化矽襯底的製造方法可以具有以下各步驟。
原料粉末準備步驟,其準備含有矽粉末、稀土元素化合物、及鎂化合物的原料粉末,其中,當將原料粉末中的矽換算成氮化矽時,原料粉末含有以氧化物計1摩爾%以上7摩爾%以下的稀土元素化合物,並含有以氧化物計8摩爾%以上15摩爾%以下的鎂化合物。
片材成形步驟,其將原料粉末成形為片狀而形成片材體。
氮化步驟,其在氮氣氛中以1200℃以上1500℃以下對片材體進行加熱,將包含在片材體中的矽氮化。
燒結步驟,其在氮氣氛下對完成了氮化步驟的片材體進行燒結。
以下對各步驟進行說明。
(原料粉末準備步驟)
在原料粉末準備步驟中,可以準備原料粉末,該原料粉末以預定比例包含在本實施方式的氮化矽襯底的製造方法中作為矽供給源的矽粉末(金屬矽粉末)、作為燒結助劑的稀土元素化合物(稀土元素化合物粉末)、及鎂化合物(鎂化合物粉末)。
如上所述在以往的氮化矽燒結體的製造方法中,提出了僅使用氮化矽粉末作為包含在起始原料中的矽供給源的方法。然而,由於氮化矽粉末昂貴,因此如果僅使用氮化矽粉末作為矽供給源則會存在製造成本上升的問題。對此,在本實施方式的氮化矽襯底的製造方法中,可以使用矽粉末作為原料,利用反應燒結來製造氮化矽襯底。
需要說明的是,作為矽供給源,除了使用矽粉末,也可以使用氮化矽粉末。換言之,作為矽供給源,可以使用矽粉末、或者矽粉末與氮化矽粉末的混合粉末。但是,在原料粉末中所包含的矽供給源之中,氮化矽粉末優選為20摩爾%以下,更優選為10摩爾%以下。進一步優選為5摩爾%以下,能夠抑制原料費。由於在原料粉末中包含的矽供給源可以僅為矽粉末,因此在原料粉末中所包含的矽供給源之中,氮化矽粉末的比例的下限值例如可以設為0摩爾%。需要說明的是,上述氮化矽的摩爾濃度是將原料粉末中的矽換算成氮化矽計算的值、也即將矽粉末的1摩爾的矽作為1/3摩爾計算時的值。具體來說,例如原料粉末包含3摩爾的矽粉末和1摩爾的氮化矽粉末作為矽供給源時,在原料粉末中所包含的矽供給源之中,包含50摩爾%的氮化矽粉末。
矽粉末、或者矽粉末與氮化矽粉末的混合粉末的平均粒徑優選在1μm以上30μm以下的範圍內,更優選在5μm以上15μm以下的範圍內。
需要說明的是,在本說明書中,平均粒徑是指通過雷射衍射散射法求出的粒度分布中積分值50%的粒徑。
在一般情況下市場銷售的氮化矽粉末或矽粉末中包含有不可避免的雜質。矽粉末中包含的氧量會隨著粉末的性狀而不同,例如在氮化矽粉末的情況中,包含1.2質量%左右,在矽粉末的情況中,包含0.2質量%至幾個質量%。
在本實施方式的氮化矽襯底的製造方法中所使用的氮化矽粉末的純度優選在98%以上99.99%以下的範圍,更優選在99%以上99.9%以下的範圍。
另外,在本實施方式的氮化矽襯底的製造方法中所使用的矽粉末的純度優選在99%以上的範圍,更優選在99.5%以上的範圍。
如下所述,在製造氮化矽襯底時,為了提高氮化矽襯底的熱導率,優選降低在氮化矽燒結體的晶體中所包含的固溶氧量。並且,根據本實施方式的氮化矽襯底的製造方法,由於是利用反應燒結並以矽粉末為起始原料,因此與作為矽供給源僅使用氮化矽作為起始原料的情況相比,從降低氧量的觀點來看具有較大的好處。在使用矽粉末作為起始原料的情況下,在將作為起始原料的原料粉末成形為片狀的片材成形步驟之後,實施將起始原料氮化的氮化步驟。並且在氮化步驟中,進行以下的(式1)所示的氮化反應。
3Si+2N2=Si3N4 (式1)
在該氮化反應中,由於試料重量增加了大約70%,因此原料粉末中的雜質氧量相對地降低。因此如上所述,通過使用矽粉末作為矽供給源,與作為矽供給源僅使用氮化矽作為起始原料的情況相比,能夠降低氮化矽燒結體的晶體中的氧量。
在本實施方式的氮化矽襯底的製造方法中如上所述通過經過氮化反應能夠相對地降低原料粉末中的雜質氧量。因此,原料的矽粉末中的雜質氧量的影響變得輕微。因此,在本實施方式的氮化矽襯底的製造方法中,可使用從雜質氧濃度較高的低品質的矽粉末到雜質氧濃度較低的高品質的矽粉末的各種矽粉末。但是,當特別需要降低氮化矽襯底中的雜質氧量時,優選使用雜質氧濃度較低的高品質的矽粉末。
在進一步對矽粉末成形體進行了氮化時,由於該成形體的重量增加而無尺寸變化,因此與氮化前的成形體相比所得到的氮化體的相對密度增高十幾%,易於在後燒結處理(燒結步驟)中的緻密化。這還具有使燒結時間的縮短成為可能、能夠防止對機械特性產生不利影響的過度的晶粒生長等優點。這樣一來,在本實施方式的氮化矽襯底的製造方法中由於利用反應燒結,因此從氮化矽襯底的高導熱化的觀點來看具有優異的特徵。
如上所述,在本實施方式的氮化矽襯底的製造方法中,在對含有矽粉末的原料粉末進行氮化的氮化步驟之後,可以經過燒結步驟來製造氮化矽襯底。然而,由於氮化矽是難燒結體,因此僅用氮化矽不進行燒結無法得到緻密體。因此,進行燒結,並為了得到緻密體而添加燒結助劑。並且,考慮(1)在氮化矽燒結體的晶體中存在固溶氧、(2)在氮化矽燒結體中存在殘留的低熱導的晶界玻璃相是燒結體的熱導率與單晶體的理論熱導率相比降低的原因。因此,通過(a)降低在氮化矽燒結體中的氮化矽顆粒中包含的固溶氧量、(b)降低氮化矽燒結體中的低熱導的晶界玻璃相,從而能夠提高氮化矽燒結體的熱導率。
因此,考慮通過以滿足上述條件的方式選擇燒結助劑,能夠達到氮化矽的高導熱化。並且,為了(a)降低在氮化矽燒結體中的氮化矽顆粒中包含的固溶氧量,優選使用與氧的親和性較高且在晶界玻璃相重捕捉氧的能力優異的稀土元素化合物作為燒結助劑。另外,為了(b)降低氮化矽燒結體中的低熱導的晶界玻璃相,優選使在加熱時生成的熔液的熔點降低、在燒結初期對緻密化做出貢獻,再有使用在高溫的燒結時蒸發揮發的鎂化合物作為燒結助劑。
然而,如上所述本發明的發明人通過對現有技術中在襯底表面產生改性層的原因進行研究,發現是由於製作氮化矽襯底時燒結助劑揮發而產生的。
因此,本發明的發明人以通過反應燒結的手法來製造高熱導率氮化矽燒結體襯底為目的,針對燒結助劑的成分對後燒結氮化體得到的氮化矽燒結體襯底的緻密化及熱導率產生的影響進行了研究。並且,發現通過使用稀土元素化合物和鎂化合物作為燒結助劑、並對其添加量進行控制從而能夠製造無需在燒結體形成後除去改性層的緻密的氮化矽襯底,並完成了本發明。
在本實施方式的氮化矽襯底的製造方法中,優選可以使用稀土元素化合物和鎂化合物作為燒結助劑。可以在原料粉末準備步驟中將用作燒結助劑的稀土元素化合物和鎂化合物與矽粉末一起作為原料粉末來準備。
對稀土元素化合物的添加量並不特別限定,當將原料粉末中的矽換算成氮化矽時,優選以原料粉末含有以氧化物計1摩爾%以上7摩爾%以下的稀土元素化合物的方式進行添加。需要說明的是,稀土元素化合物的添加量(摩爾濃度)是指原料粉末中所包含的矽供給源(包括矽粉末、有時進一步包括氮化矽粉末)、稀土元素化合物、及鎂化合物三個成分中的稀土元素化合物的摩爾濃度,以下的說明中也同樣。
如上所述稀土元素化合物與氧的親和性較高、在晶界玻璃相中捕捉氧的能力優異。因此,當稀土元素化合物的添加量小於1摩爾%時,在晶界相(晶界玻璃相)中無法捕捉氧,在氮化矽顆粒中所包含的固溶氧量變多,熱導率變低。另外,如果超過7摩爾%,則包含稀土元素化合物的低熱導的晶界相的量有時會變多,燒結體的熱導率有時會降低。
特別是,當將原料粉末中的矽換算成氮化矽時,更優選以原料粉末含有以氧化物計1.5摩爾%以上5摩爾%以下的稀土元素化合物的方式進行添加,進一步優選以原料粉末含有以氧化物計2摩爾%以上4摩爾%以下的稀土元素化合物的方式進行添加。
作為稀土元素化合物並無特別限定,包含在稀土元素化合物中的稀土元素優選包括選自Y、Sc、La、Ce、Nd、Sm、Gd、Dy、Ho、Er、Yb的一種以上的元素。特別是,包含在稀土元素化合物中的稀土元素更優選為選自Y、Sc、La、Ce、Nd、Sm、Gd、Dy、Ho、Er、Yb的一種以上的元素。作為稀土元素化合物,例如可舉出稀土元素氧化物。作為稀土元素氧化物,具體例如可以優選使用氧化釔、氧化鈰,氧化鐿或氧化鈧等。需要說明的是,稀土元素化合物並不限定於一種,可以同時使用兩種以上的稀土元素化合物。
對於鎂化合物的添加量並無特別限定,當將原料粉末中的矽換算成氮化矽時,優選含有以氧化物計8摩爾%以上15摩爾%以下的鎂化合物。需要說明的是,鎂化合物的含量(摩爾濃度)是指包含在原料粉末中的矽供給源(包括矽粉末,有時進一步包括氮化矽粉末)、稀土元素化合物、及鎂化合物三種成分中的鎂化合物的摩爾濃度,以下的說明也相同。
當僅添加稀土元素化合物作為燒結助劑來製作氮化矽燒結體時,為了進行緻密化需要10MPa左右的高氮壓中、高達2000℃的超高溫下的燒結,由於需要特殊的燒結爐因此處理成本變高。另外,由於超高溫下的燒結而產生顯著的晶粒生長,招致機械特性的降低。因此,為了能夠實現高強度、高韌性,優選在添加稀土元素化合物的同時、添加鎂化合物。通過添加鎂化合物,有使鎂離子變成加熱時生成的氮氧共滲玻璃的修飾離子、降低玻璃的粘性、促進緻密化、同時在燒結中進行蒸發和揮發、降低殘留的晶界相的量的作用。並且,當鎂化合物的添加量小於8摩爾%時,在進行燒結收縮前鎂會揮發,因此產生改性層、無法得到緻密體。另外,當鎂化合物的添加量大於15摩爾%時,有可能在後燒結(燒結步驟)後也會殘留大量的鎂、阻礙燒結體的熱導率。因此,如上所述當將原料粉末中的矽換算成氮化矽時,優選以含有以氧化物計8摩爾%以上15摩爾%以下的鎂化合物的方式進行添加。另外,當將原料粉末中的矽換算成氮化矽時,更優選以含有以氧化物計8摩爾%以上10摩爾%以下的鎂化合物的方式添加鎂化合物。
作為鎂化合物並無特別限定,例如可以使用鎂的矽化物、氟化物、硼化物、氮化物、以及這些的三元化合物。特別是,從使用的容易性、處理時的穩定性、不會產生有害物質等的觀點來看,添加在原料粉末中的鎂化合物優選包含選自氧化鎂(MgO)、矽化鎂(Mg2Si)、或氮化矽鎂(MgSiN2)的一種以上的鎂化合物。另外,添加在原料粉末中的鎂化合物優選是選自氧化鎂(MgO)、矽化鎂(Mg2Si)、或氮化矽鎂(MgSiN2)的一種以上的鎂化合物。
在如上所述的原料粉末準備步驟中準備的原料粉末也可以為將作為矽供給源的矽粉末、稀土元素化合物、及鎂化合物混合的混合粉末。需要說明的是,原料粉末如上所述有時可以進一步包含氮化矽作為矽供給源。
另外,也可以對原料粉末中所包含的成分(粉末)進行稱量後不進行混合,在下述的片材成形步驟中在用於形成片材體而形成漿料時與構成漿料的其他成分一併混合、粉碎。
(片材成形步驟)
在片材成形步驟中,可以將以在原料粉末準備步驟中為預定成分的方式所準備的原料粉末成形為片狀而形成片材體(生胚片材體)。
具體來說,首先,對於在原料粉末準備步驟中以設為預定成分的方式準備的原料粉末,可以使用水或有機溶劑作為分散介質,根據需要添加有機類粘合劑(有機類粘結劑)或分散劑,通過球磨機或行星式磨機利用通常方法進行混合。
需要說明的是,如上所述在原料粉末準備步驟中,可以將在原料粉末中所包含的矽粉末(有時進一步包含氮化矽粉末)、稀土元素化合物、或鎂化合物等預先混合為混合粉末,將該混合粉末與上述分散介質等混合。另外,可以在原料粉末準備步驟中,僅對在原料粉末中所包含的矽粉末等進行稱量,向放入有上述分散介質等的球磨機中投入在原料粉末中所包含的矽粉末等各種粉末,同時實施原料粉末的混合、以及原料粉末與分散介質等的混合。
在片材成形步驟中,對於片材成形的試料進行調試時添加的分散介質、有機類粘合劑、分散劑的材料、添加量、或添加方法並無特別限定,可以根據片材成形方法等任意地選擇。
以下針對向原料粉末添加分散介質、有機類粘合劑、分散機的具體條件例進行說明。
在原料粉末準備步驟中準備以所說明的預定比例稱量各原料粉的原料粉末。接著,例如向加入、混合有相對於原料粉末的0.5重量%以上2重量%以下的分散劑及30重量%以上70重量%以下的有機溶劑等分散介質的球磨機等的研磨容器中添加原料粉末。作為分散劑例如可使用山梨糖醇酯型或聚氧化烯型等,作為分散介質的有機溶劑例如可使用乙醇或甲苯等。需要說明的是,如上所述作為分散介質也可以使用水。另外,可以不添加分散劑,僅使用分散介質。
接著,例如可以利用球磨機進行原料粉末的混合粉碎。對於混合粉碎的進行時間,由於其隨著所使用的研磨裝置、或起始原料的量、特性而不同因而並未特別限定,優選以能夠充分粉碎、混合原料粉末的方式來選擇時間。粉碎混合時間例如優選6小時以上48小時以下,更優選12小時以上24小時以下。例如當粉碎混合時間小於6小時時,有時燒結助劑未被均勻混合而在燒結中產生不均、或者有時矽粉末依舊以粗大地形式存在、會對絕緣特性等產生影響。另外,即使進行混合粉碎超過48小時,混合狀態也不會發生較大變化,而且有可能會從球或鍋混入雜質。需要說明的是,粉碎混合後,也可以根據需要將分散介質除去。
也可以在粉碎混合之後進一步添加5重量%以上30重量%以下的有機類粘合劑(有機類粘結劑)進行混合製作漿料。對於有機類粘合劑也並無限定,例如可以優選使用PVB類(聚乙烯醇縮丁醛樹脂)、乙基纖維素類樹脂、或丙烯酸樹脂等。
對於添加有機類粘合劑後的混合時間,由於其隨著研磨裝置等用於混合的裝置的性能等而不同因而並無特別限定,例如優選為1小時以上24小時以下,更優選6小時以上12小時以下。當混合時間小於1小時時,在未將有機類粘合劑與原料粉末均勻混合而進行片材成形時,有時所製作的片材上會產生裂紋因此不優選。另外,通常,即使進行混合超過24小時有機粘合劑與原料粉末的混合狀態也不會發生較大變化,因此從生產性的觀點來看優選混合時間為24小時以下。
在添加並混合有機類粘合劑後,可以對所製作的漿料進行真空消泡並調節漿料的粘度,用片材成形機進行塗布,進行片材成形。
如上所述利用球磨機或行星磨機將原料粉末和分散介質等混合後,可以根據需要除去分散介質,根據情況進一步添加有機類粘合劑後,將原料粉末、或包含原料粉末的漿料成形為片狀而形成片材體。對於將原料粉末成形為片狀的方法並無特別限定,例如可使用模具成形、片材成形、擠壓成形、淨水壓加壓成形(CIP成形)等。在本實施方式的氮化矽襯底的製造方法中由於並未形成改性層、無需進行除去,因此無需考慮改性層部分的厚度。因此,可以優選使用能夠成形為較薄的片狀的片材成形。
對於在片材成形步驟中形成的片材體的形狀、尺寸並無特別限定,可以根據作為氮化矽襯底時所要求的形狀、尺寸設為任意的形狀、尺寸。例如在成形步驟中形成的片材體的厚度優選為0.05mm以上2.5mm以下,更優選為0.25mm以上1.0mm以下。
對於在片材成形步驟中得到的片材體,可以根據需要例如用衝壓機等切割成預定的大小。
在片材成形步驟中,所得到的片材體的相對密度優選為45%以上,更優選為50%以上。需要說明的是,當如上所述對所得到的片材體進行切割時,優選切割後的片材體的相對密度滿足上述範圍。在片材成形步驟中,可以通過在片材成形步驟供給的漿料中所包含的原料粉末的量(固體濃度)和在漿料中添加的粘合劑量來調節所得到的片材體的相對密度。通過使在片材成形步驟中所得到的片材體的相對密度為45%以上,從而能夠充分減少片材體內的孔隙,能夠進一步提高在下述燒結步驟後所得到的氮化矽襯底的相對密度。需要說明的是,對於在片材成形步驟中所得到的片材體的相對密度的上限值並無特別限定,為了提高相對密度需要減少粘合劑等的添加量並提高漿料的固體濃度,但由於會產生裂紋等難以處理因此例如優選65%以下,更優選60%以下。
(氮化步驟)
在氮化步驟中,通過在氮氣氛中對在片材成形步驟中形成的片材體進行加熱,從而能夠氮化片材體中所包含的矽。
需要說明的是,在上述片材成形步驟中對片材體成形後進行上述氮化之前,為了除去在片材體中所包含的用於成形的有機粘合劑等,也可以在800℃以下的溫度下進行預燒(脫粘合劑步驟)。
另外,為了除去在開始氮化步驟前在爐內存在的氣體,優選暫且將爐內抽成真空並向爐內供給氮氣,並開始氮化步驟。對於在供給氮氣前將爐內抽成真空的程度並無特別限定,例如優選抽真空到1Pa以下,更優選抽真空到10-1Pa以下。
對於氮化步驟中的加熱溫度並無特別限定,例如優選為1200℃以上1500℃以下,更優選為1350℃以上1480℃以下。
對於氮化步驟的時間,優選在1小時以上15小時以下的範圍,進一步優選在3小時以上10小時以下的範圍。
這是因為例如當氮化步驟中的加熱溫度小於1200℃時、或者當氮化步驟的時間過短時,有時在片材體中會殘留未反應的矽粉末、燒結步驟後無法得到緻密體。另外,因為當以高於1500℃的溫度進行氮化反應時、或氮化步驟的時間過長時,有時燒結助劑成分會揮發、在燒結步驟中燒結助劑成分會不足,變得難以得到燒結後緻密體。
對於在氮化步驟中加熱片材體的方法並無特別限定,例如可以將片材體置於剝離用的BN(氮化硼)粉或BN板之間並堆疊,設置在由石墨隔熱材料和石墨加熱器構成的真空加壓氣氛爐中來進行實施。真空加壓氣氛爐通過使用例如緊箱式電爐來將在內部產生的氣體排放到外部。通過利用能夠將內部產生的氣體排放到外部的爐、具體來說例如緊箱式電爐,從而能夠利用一個爐來實施以下步驟,例如還實施將用於成形的有機類粘合劑除去的脫粘合劑步驟時的脫粘合劑步驟、氮化步驟、以及燒結步驟。因此,由於能夠提高生產性,因此優選。
(燒結步驟)
在燒結步驟中,可以在氮氣氛下對進行了氮化步驟的片材體進行燒結。
對於燒結步驟中的加熱溫度並無特別限定,例如優選以1700℃以上1950℃以下來進行加熱,更優選以1750℃以上1900℃以下來進行加熱。
對於燒結步驟的時間,優選在1小時以上48小時以下的範圍,進一步優選在5小時以上24小時以下的範圍。
這是因為,當作為後燒結溫度的燒結步驟的加熱溫度小於1700℃時、或燒結步驟的時間過短時,有時無法充分地將片材體緻密化。另一方面,因為當燒結步驟的加熱溫度為高於1950℃的溫度時、或燒結步驟的時間過長時,有時會產生過度的晶粒生長、所得到的氮化矽襯底的強度會降低。
優選在燒結步驟中在氮氣氛下進行加熱,但對此時的氮氣氛壓力並無特別限定,例如優選通過燒結步驟的加熱溫度在不使氮化步驟中生成的氮化矽分解的程度的壓力下對其進行加熱。具體來說例如優選為0.1MPa以上,更優選為0.9MPa以上。但是,如果氮氣氛的壓力變得過高,則由於產生使用耐壓性較高的特殊的爐的必需性,因此例如氮的氣氛壓力優選為1MPa以下,更優選為0.92MPa以下。
通過實施燒結步驟,從而能夠得到例如相對密度為95%以上的氮化矽襯底,能夠使在燒結步驟後得到的氮化矽襯底為不含改性層的緻密的襯底。因此,對於在燒結步驟後所得到的氮化矽襯底,在未加工的狀態下,利用雷射閃光法測定的熱導率可以為80W/mK以上,優選可以為110W/mK以上。
另外,對於實施了燒結步驟的氮化矽襯底,可以為以β相氮化矽為主成分、含有稀土元素的襯底。需要說明的是,稀土元素可以是單體的狀態,也可以與其他物質形成化合物。此時,在氮化矽襯底中,優選包含以氧化物計1摩爾%以上4摩爾%以下的稀土元素。另外,對於實施了燒結步驟的氮化矽襯底的鎂的存在量,優選為以氧化物計2摩爾%以下。當實施了燒結步驟的氮化矽襯底含有鎂時,鎂可以為單體的狀態,也可以與其他物質形成化合物。
對於燒結步驟後所得到的氮化矽襯底的厚度並無特別限定,可以為任意的厚度,例如當用作半導體元件或電子設備的絕緣散熱襯底時,優選為0.05mm以上2.5mm以下。需要說明的是,可以通過對在片材成形步驟中形成的片材體的厚度進行調整來選擇在燒結步驟後所得到的氮化矽襯底的厚度。
根據以上說明的本實施方式的氮化矽襯底的製造方法,能夠提供一種氮化矽襯底的製造方法,其使用包含矽粉末的原料粉末,並且氮化矽襯底的表面不存在改性層、被緻密化成95%以上的相對密度。並且,根據本實施方式的氮化矽襯底的製造方法,能夠提供一種氮化矽襯底的製造方法,該氮化矽襯底在燒結後未加工的狀態下,利用雷射閃光法測定的熱導率為80W/mK以上,優選為110W/mK以上。
換言之,根據本實施方式的氮化矽襯底的製造方法,能夠提供一種緻密的高導熱性氮化矽襯底的製造方法,其能夠利用反應燒結的手法進行合成,具有較高的可靠性,在形成燒結體後無需除去改性層。再有,能夠提供一種利用該製造方法製造的高導熱性氮化矽襯底及其應用產品。
另外,由於在利用本實施方式的氮化矽襯底的製造方法所得到的氮化矽襯底中如上所述在表面不存在改性層,因此變得無需像現有技術那樣預先製造較厚的氮化矽襯底,將改性層部分除去。因此,與現有技術相比能夠消減步驟個數,降低成本。再有,由於向氮化步驟或燒結步驟提供的片材體也可以較薄,因此能夠在片材體的成形中利用薄板成形法、例如片材成形等。
<實施例>
以下舉出具體的實施例來進行說明。本發明不限定於該實施例。
[實施例1]
按照以下步驟製造樣品編號1-1~1-4四種氮化矽襯底。
(原料粉末準備步驟)
作為矽粉末,使用純度99.9%、平均粒徑10μm、雜質氧量0.10質量%的粉末。作為鎂化合物,使用平均粒徑0.1μm的氧化鎂粉末(宇部材料株式會社制)或平均粒徑1.0μm的氮化矽鎂粉沫。另外,作為稀土元素化合物,使用平均粒徑1.5μm的氧化釔粉末(信越化學工業株式會社制)。
需要說明的是,對於矽粉末的雜質氧量,使用氮氧同時分析裝置(LECO日本公司制。型號:TC-600)進行測定。
對於上述原料按表1所示的樣品編號1-1~1-4的各個成分來稱量,並準備原料粉末。需要說明的是,表1中僅表示了鎂化合物和稀土元素化合物的比例,剩餘部分為矽粉末。另外,表1所示的摩爾比表示當假定將矽(Si)完全氮化成氮化矽(Si3N4)、將矽換算成氮化矽、將鎂化合物換算成氧化鎂(MgO)時的摩爾比。
(片材形成步驟)
使用乙醇作為分散介質,使用樹脂鍋和氮化矽球,對在原料粉末準備步驟中所準備的各樣品的原料粉末用球磨機進行粉碎混合24小時。需要說明的是,對於乙醇以漿料濃度為45重量%的方式預先稱量,投入樹脂鍋內。粉碎混合後,添加作為有機類粘合劑的樹脂粘合劑(積水化學工業,商品名「S-LEC」)10重量%,進一步混合12小時。接著,使用真空脫泡機(Sayama理研製)進行粘度調節,製作塗布用漿料。對於進行了粘度調節的漿料,使用刮刀將各樣品片材成形為片材厚度0.4mmt。片材成形後,在切割為40×40×0.4mmt後,對片材的相對密度進行評價。相對密度的評價是通過測長來進行。關於所得到的片材的相對密度,各樣品均為53.0%~54.8%。
(氮化步驟)
在片材成形步驟中對片材體進行成形、評價後,在片材體的表面上塗布氮化硼粉末(以下也記載為「BN粉末」),以12片為1組進行層疊,設置在氮化硼制坩堝(以下也記載為「BN坩堝」)中。之後,將BN坩堝設置在真空加壓氣氛爐(富士電波工業株式會社制。型號:Multi500)中,在真空中以800℃加熱4小時,進行脫粘合劑步驟。脫粘合劑步驟結束後,將爐內抽真空至10-1Pa,向爐內倒入氮,並在0.1MPa的氮氣氛中以1400℃進行氮化處理8小時。作為氮氣使用99.9體積%的氮氣。
當對所得到的氮化體進行了X射線衍射測定(株式會社Rigaku制。型號:RINT2500)後,對於各個試料均未確認到殘留Si。
(燒結步驟)
接著,作為後燒結,對於在氮化步驟中對各樣品實施了氮化處理的片材體按照表1所示的條件進行燒結。需要說明的是,與氮化步驟相同,燒結步驟使用真空加壓氣氛爐,同樣地將層疊的片材體設置在BN坩堝中,進一步將BN坩堝設置在真空加熱氣氛爐內進行實施。
燒結步驟後,從BN坩堝中取出燒結的片材體(氮化矽襯底),用噴砂裝置將附著在表面上的BN粉末等除去。對由此得到的襯底,利用X射線衍射進行相鑑定,使用阿基米德法對相對密度進行測定。另外,利用掃描式電子顯微鏡(SEM)(日本電子株式會社制。型號:JSM-5600)對所得到的氮化矽襯底的剖面進行觀察。
再有,將所得到的氮化矽襯底切割為25mm×25mm的方形,利用雷射閃光法(ULVAC株式會社制。型號:TC-9000)對熱導率進行測定。
表1中表示出評價結果。另外,圖1中表示出樣品編號1-1的剖面的SEM的觀察結果。
[表1]
首先,通過X射線衍射進行相鑑定,能夠確認對於各個樣品都得到了氮化矽。
如圖1所示的製作的樣品編號1-1的氮化矽襯底的剖面也顯而易見,當進行SEM觀察後,能夠確認不存在改性層。需要說明的是,對於本實施例的其他樣品同樣地進行了觀察後,也能夠確認不存在改性層。
另外,根據表1所示的結果顯而易見,能夠確認實施例1中製作的氮化矽襯底均是相對密度為95%以上的緻密體,對於熱導率最低的試料也為110W/mK、最高的試料為149W/mK。
[實施例2]
在原料粉末準備步驟中,作為矽粉末,使用純度99.5%的粉末。以表1所示的樣品編號1-5~1-8的各成分進行稱量,準備原料粉末。此外,按照與實施例1相同的條件製作實施例2的氮化矽襯底。能夠確認實施例2中所製作的氮化矽襯底均得到了氮化矽、並且不存在改性層。再有,能夠確認實施例2中所製作的氮化矽襯底均是相對密度為99%以上的緻密體,熱導率最低的試料也為99W/mK、最高的試料為133W/mK。
[實施例3]
在原料粉末準備步驟中,作為矽粉末,使用純度99%的粉末。以表1所示的樣品編號1-9~1-12的各成分進行稱量,準備原料粉末。此外,按照與實施例1相同的條件製作實施例3的氮化矽襯底。能夠確認實施例3中所製作的氮化矽襯底均得到了氮化矽、並且不存在改性層。能夠確認實施例3中所製作的氮化矽襯底均是相對密度為99%以上的緻密體,熱導率最低的試料也為80W/mK、最高的試料為116W/mK。
[比較例1]
按照以下步驟製作氮化矽襯底,進行評價。
作為矽粉末,使用純度99.9%、平均粒徑10μm、雜質氧量0.16質量%的粉末。另外,作為鎂化合物,使用平均粒徑0.1μm的氧化鎂粉末(宇部材料株式會社制)。作為稀土元素化合物,使用平均粒徑1.5μm的氧化釔粉末(信越化學工業株式會社制)。需要說明的是,對於矽粉末的雜質氧量,與實施例1的情況同樣,使用氮氧同時分析裝置進行測定。
以調配上述原料粉末時的摩爾比,按Si3N4:Y2O3:MgO=93:2:5來進行稱量。需要說明的是,上述摩爾比表示假定將矽(Si)完全氮化為氮化矽(Si3N4)(也即將矽換算成氮化矽)、將鎂化合物換算成氧化鎂(MgO)時的摩爾比。
對稱量了上述成分的原料粉末,使用行星磨機(Fritsch日本株式會社制)在乙醇中以轉動速度250rpm的條件混合2小時。需要說明的是,在混合時使用氮化矽制鍋及氮化矽制球(直徑φ1.0mm)。
對於與乙醇混合的原料粉末,用蒸發器使溶劑揮發,以110℃進行真空乾燥4小時得到作為起始原料的原料粉末。通過模具擠壓成型將所得到的大約36g的原料粉末成形為75×75×5mm的尺寸後,以300MPa進行CIP處理。
在BN坩堝中將成形體埋入BN粉末,進一步將BN坩堝設置在碳坩堝中。接著,使用碳加熱器爐,在0.1MPa的氮中以1400℃進行氮化8小時。接著,在氮化後,在0.9MPa的加壓氮中以1900℃進行後燒結6小時。
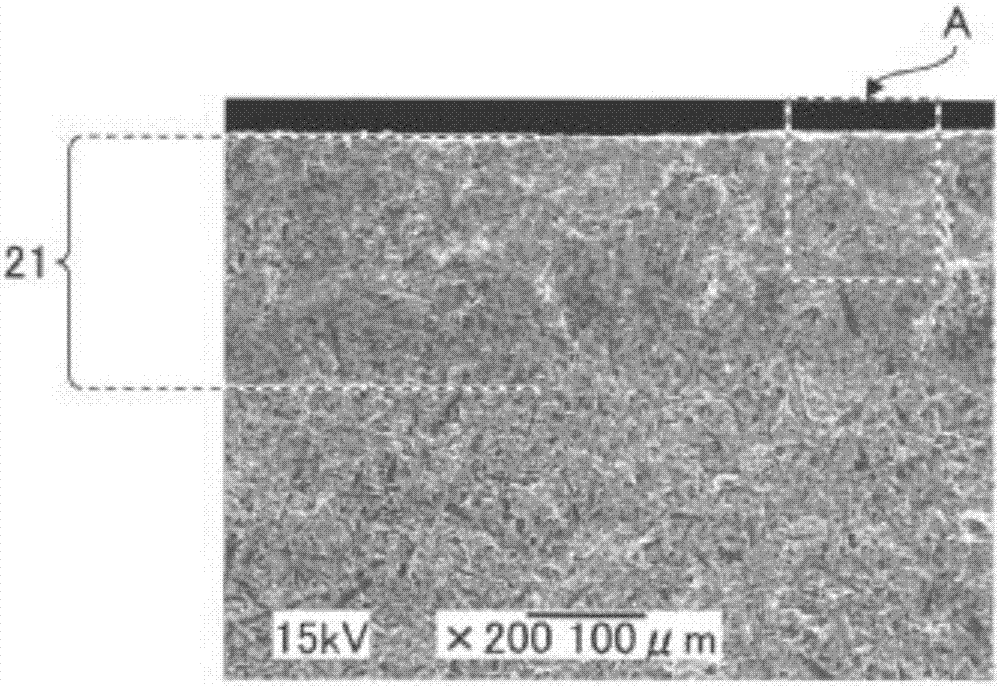
對於所得到的燒結體,利用阿基米德法測定相對密度。另外,與實施例1同樣地對剖面進行SEM觀察。結果表示在圖2A、圖2B中。需要說明的是,圖2B相當於由圖2A的虛線A所包圍的區域的放大圖。
對於所製作的試料,如圖2A、圖2B所示,能夠確認在表層部產生了含有多個氣孔的改性層21,改性層21的厚度大約為200μm。
另外,對於所得到的氮化矽襯底,相對密度為98.7%,如上所述在表層部包含改性層,為了用作襯底需要除去改性層。
[比較例2]
按照以下步驟製作樣品編號2-1、2-2兩種氮化矽襯底,進行評價。
作為矽粉末,使用純度99.9%、平均粒徑10μm、雜質氧量0.16質量%的粉末。另外,作為鎂化合物,使用平均粒徑0.1μm的氧化鎂粉末(宇部材料株式會社制)或平均粒徑1.0μm的氮化矽鎂粉末。另外,作為稀土元素化合物,使用平均粒徑1.5μm的氧化釔粉末(信越化學工業株式會社制)。需要說明的是,對於矽粉末的雜質氧量,使用氮氧同時分析裝置進行測定。
按表2所示的成分稱量上述原料準備原料粉末。需要說明的是,表2中僅表示出鎂化合物和稀土元素化合物的比例,剩餘部分為矽粉末。另外,表2所示的摩爾比表示假定將矽(Si)完全氮化為氮化矽(Si3N4)、將矽換算成氮化矽、將鎂化合物換算成氧化鎂(MgO)時的摩爾比。
以甲醇作為分散介質,使用樹脂鍋和氮化矽球,用球磨機對上述原料粉末進行粉碎混合24小時。粉碎混合後,添加10質量%的作為有機類粘合劑的樹脂粘合劑(積水化學工業,商品名「S-LEC」),混合12小時。使用真空脫泡機進行粘度調節,製作各樣品的塗布用漿料。對於粘度調節了的漿料,使用刮刀片材成形為片材厚度0.4mmt。
片材成形後,切割成40×40×0.4mmt後,對片材的相對密度進行評價。需要說明的是,相對密度的評價是通過測長來進行。所得到的片材的相對密度為53.6%。
之後,在片材體的表面塗布BN粉末,以12片為1組進行層疊,設置在BN坩堝中。接著,將BN坩堝設置在真空加壓氣氛爐中,在真空中以800℃加熱4小時,進行脫粘合劑處理。
接著,將真空加壓氣氛爐內暫且抽真空至1Pa以下後、向爐內導入氮,在0.11MPa的氮中以1450℃加熱4小時,進行氮化處理。當對氮化處理後所得到的樣品進行X射線衍射測定後,在各個樣品中均未確認到殘留Si。
接著,作為後燒結,在0.9MPa的加壓氮中以1900℃對氮化體燒結6小時。
燒結步驟後,從BN坩堝中取出板狀燒結體。
[比較例3]
除了使用純度99%的矽粉末,按照與樣品編號2-1及2-2分別同樣的步驟製造樣品編號2-3及2-4的氮化矽襯底,並進行評價。
對於所得到的襯底,使用阿基米德法測定相對密度,利用X射線衍射進行相鑑定。另外,利用掃描式電子顯微鏡(SEM)(日本電子株式會社制。型號:JSM-5600)對所得到的氮化矽襯底的剖面進行觀察。結果表示在表2、圖3中。需要說明的是,圖3表示出利用SEM對樣品編號2-1的剖面進行觀察的結果。
[表2]
首先,當利用X射線衍射進行相鑑定後,能夠確認在各個樣品中均得到了氮化矽。
另外,能夠確認如表2所示製作的氮化矽襯底的相對密度降低為67.2%、68.4%。考慮這是因為如圖3所示對於襯底整體變成了包含多個氣孔的多孔隙的改性層31,在本比較例中未能得到緻密體。在圖3所示的襯底中,由於襯底整體變成了改性層31,因此改性層31的厚度為400μm。
需要說明的是,圖3是對樣品編號2-1的剖面進行觀察的SEM圖像,對於另外的樣品編號2-2~2-4也能夠同樣地確認襯底整體變成了改性層。
以上通過實施方式及實施例等對氮化矽襯底的製造方法進行了說明,但本發明並不限定上述實施方式及實施例等。在權利要求書中所記載的本發明的主旨的範圍內,可進行各種變形、變更。
本申請以在2014年3月31日向日本特許廳申請的日本專利申請第2014-074340號、及在2015年3月31日向日本特許廳申請的日本專利申請第2015-072347號作為要求優先權的基礎,本國際申請援引日本專利申請第2014-074340號、及日本專利申請第2015-072347號的全部內容。