一種分離富集全氟辛烷磺酸的磁性分子印跡聚合物及其製備方法與流程
2023-04-23 18:27:36 2
本發明屬於磁性分子印跡聚合物技術領域,具體涉及一種分離富集全氟辛烷磺酸(perfluorooctanesulfonate,pfos)的磁性分子印跡聚合物及其製備方法。
背景技術:
近年來,pfos被廣泛使用在工業和消費領域,但進一步研究表明,pfos對動物及人具有系統性毒性,例如神經毒性、胚胎發育和生殖毒性以及致癌性等。2009年,pfos被正式列入《斯德哥爾摩公約》的附件b中,該公約限制了全球對pfos的生產和使用。目前處理pfos主要的方法包括零價鐵還原、超聲降解、光催化以及膜分離等,但其需要較高的消費及能量消耗。然而,物理吸附劑包括活性炭、有機膨潤土、鈦酸納米管等雖然在一定程度上能有效去除pfos,但其選擇性低、重複利用率低、水處理設備昂貴、耗時等缺點。
分子印跡技術(molecularimprintingtechnique,mip)是以某一特定目標分子為模板,製備對該分子具有特異選擇性聚合物的過程,其聚合物表面的結合位點與模板分子在形狀、尺寸及空間結構上能很好的匹配,通常被描述為「分子鑰匙」的人工「鎖」技術,但由於其低傳質、低結合能、慢結合動力學、分離困難等缺點,難以實際應用,如文獻waterres.2008,42:3089~3097,其用殼聚糖合成的pfos分子印跡,不僅難以分離,且吸附動力學時間長達32h。磁性分離技術是以納米或微粒級的磁性微粒為載體,在外加磁場的定向控制下,通過吸附、清洗、解吸操作,能夠在便於分離的基礎上,降低傳質阻力、提高吸附選擇性,成為本領域研究的熱點。
技術實現要素:
本發明的一個目的是提供一種分離富集全氟辛烷磺酸的磁性分子印跡聚合物,所述聚合物具有超薄的分子印跡薄膜、快速的吸附動力學、高的吸附選擇性和重複使用性,能夠有效、快速去除水中的全氟辛烷磺酸。
本發明的第二個目的是提供上述分離富集全氟辛烷磺酸的磁性分子印跡聚合物的製備方法。
為實現上述發明目的,提供以下技術方案:
一種分離富集全氟辛烷磺酸的磁性分子印跡聚合物,其特徵在於:以fe3o4@sio2@ch=ch2磁性納米粒為核,全氟辛烷磺酸分子印跡聚合物為殼,由以下方法製備得到:
步驟(1)將全氟辛烷磺酸和丙烯醯胺按質量比1:5~30的比例加入乙腈中,常溫下攪拌12h,形成預組裝溶液;
步驟(2)向步驟(1)所得的預組裝溶液中依次加入乙二醇二甲基丙烯酸酯、偶氮二異丁腈和fe3o4@sio2@ch=ch2,氮氣保護下,60℃下攪拌8~30h,分離,除去模板分子,其中,全氟辛烷磺酸、丙烯醯胺、乙二醇二甲基丙烯酸酯、偶氮二異丁腈和fe3o4@sio2@ch=ch2的質量比為1:(5~30):79.3:4.5:(5~20)。
根據本發明所述的磁性分子印跡聚合物,步驟(1)中所述fe3o4@sio2@ch=ch2由以下方法製備得到:
步驟a將fecl3·6h2o溶解於乙二醇中,形成黃色的透明溶液,依次加入無水醋酸鈉和1,6-己二胺,50℃機械攪拌30min,轉入高溫反應釜在198℃反應6h,磁性分離,超純水和乙醇洗滌,真空乾燥,其中,fecl3·6h2o和無水醋酸鈉的質量比為1:2;
步驟b將步驟a所得物分散乙醇/水(體積比4:1)溶液中,超聲15min,加入nh3·h2o和原矽酸四乙基酯(teos),40℃機械攪拌12h,磁性分離,超純水和乙醇洗滌,真空乾燥,其中,每克步驟a所得物使用16.7毫升nh3·h2o和2.3毫升teos;
步驟c將步驟b所得物分散於質量分數為10%的醋酸溶液中,加3-(異丁烯醯氧)丙基三甲氧基矽烷(mps),60℃機械攪拌5h,磁性分離,用超純水和乙醇洗滌,真空乾燥。
具體地,根據本發明所述的磁性分子印跡聚合物,步驟(1)中所述fe3o4@sio2@ch=ch2由以下方法製備得到:
步驟a將fecl3·6h2o溶解於乙二醇中,形成黃色的透明溶液,依次加入無水醋酸鈉和1,6-己二胺,50℃機械攪拌30min,轉入高溫反應釜在198℃反應6h,磁性分離,超純水和乙醇洗滌,真空乾燥,其中,fecl3·6h2o和無水醋酸鈉的質量比為1:2,乙二醇的用量為每克fecl3·6h2o使用30毫升乙二醇和7.6毫升1,6-己二胺;
步驟b將步驟a所得物分散乙醇/水(體積比4:1)溶液中,超聲15min,加入nh3·h2o和原矽酸四乙基酯(teos),40℃機械攪拌12h,磁性分離,超純水和乙醇洗滌,真空乾燥,其中,每克步驟a所得物使用16.7毫升nh3·h2o和2.3毫升teos;
步驟c將步驟b所得物分散於質量分數為10%的醋酸溶液中,加3-(異丁烯醯氧)丙基三甲氧基矽烷(mps),60℃機械攪拌5h,磁性分離,用超純水和乙醇洗滌,真空乾燥,其中,每克步驟b所得物使用0.6毫升mps。
根據本發明所述的磁性分子印跡聚合物,步驟(1)中所述全氟辛烷磺酸與丙烯醯胺的質量比為1:14.22。
根據本發明所述的磁性分子印跡聚合物,步驟(1)中所述的攪拌為機械攪拌。
根據本發明所述的磁性分子印跡聚合物,步驟(1)中所述乙腈的用量為3毫升每毫克全氟辛烷磺酸。
根據本發明所述的磁性分子印跡聚合物,步驟(2)中所述氮氣保護為向溶液中通入氮氣10~60min;優選地,所述氮氣保護為向溶液中通入氮氣30min。
根據本發明所述的磁性分子印跡聚合物,步驟(2)中全氟辛烷磺酸、丙烯醯胺、乙二醇二甲基丙烯酸酯、偶氮二異丁腈和fe3o4@sio2@ch=ch2的質量比為1:14.22:79.3:4.50:10。
根據本發明所述的磁性分子印跡聚合物,步驟(2)中所述的攪拌為機械攪拌24h。
根據本發明所述的磁性分子印跡聚合物,步驟(2)中所述的分離為磁性分離。
根據本發明所述的磁性分子印跡聚合物,步驟(2)中所述的除去模板分子的方法是用甲醇/乙酸(v:v=9:1)混合溶液除去;優選地,步驟(2)中所述的除去模板分子的方法是將磁性分離的聚合物用乙腈洗滌至上清液澄清後,用甲醇/乙酸(v:v=9:1)混合溶液除去模板分子,甲醇洗滌至溶液呈中性,磁性分離,真空乾燥。
本發明人在實驗中發現,製備的磁性分子印跡聚合物的性質與核的材料和製備方法、pfos與丙烯醯胺的質量比等因素密切相關。當pfos與丙烯醯胺形成的複合物在以fe3o4@sio2@ch=ch2為核、乙二醇二甲基丙烯酸酯為交聯劑、偶氮二異丁腈為引發劑時,可誘導雙鍵間發生聚合反應,從而形成空間網狀結構。在洗脫模板分子後,磁性分子印跡聚合物表面形成的結合位點在形狀、尺寸、空間結構與模板分子能夠較好的匹配,故特異選擇性好。另外,由於形成的磁性分子印跡聚合物體積小,因此比表面積增大,結合位點數量增多,最終吸附量增大;製備過程中pfos與丙烯醯胺的質量比對磁性分子印跡聚合物對pfos的吸附量有顯著影響,當丙烯醯胺的量過少量時,形成的分子印跡聚合物結合位點較少,吸附量降低,全氟辛烷磺酸與丙烯醯胺的質量比為1:5~30,尤其是1:14.22時,效果最佳。
此外,本發明人還發現,pfos溶解性較差,不利於模板分子與功能單體相互作用,導致合成的聚合物對pfos的吸附量降低,故本發明通過先將pfos與丙烯醯胺攪拌12小時,使模板與功能單體之間通過靜電等相互作用預組裝,形成複合物,有利於磁性分子印跡聚合物的形成。
第二方面,本發明提供上述分離富集全氟辛烷磺酸的磁性分子印跡聚合物的製備方法,包括以下步驟:
步驟(1)將全氟辛烷磺酸和丙烯醯胺按質量比1:5~30的比例加入乙腈中,常溫下攪拌12h,形成預組裝溶液;
步驟(2)向步驟(1)所得的預組裝溶液中依次加入乙二醇二甲基丙烯酸酯、偶氮二異丁腈、fe3o4@sio2@ch=ch2,氮氣保護下,60℃下攪拌8~30h,磁性分離,除去模板分子,其中,全氟辛烷磺酸、丙烯醯胺、乙二醇二甲基丙烯酸酯、偶氮二異丁腈和fe3o4@sio2@ch=ch2的質量比為1:(5~30):79.3:4.5:(5~20)。
在一些優選的實施方案中,一種用於分離富集全氟辛烷磺酸的磁性分子印跡聚合物的製備方法,包括以下步驟:
步驟(1):將全氟辛烷磺酸和丙烯醯胺按質量比1:14.22的比例加入乙腈中,常溫下機械攪拌12h,形成預組裝溶液,所述乙腈的用量為3毫升每毫克全氟辛烷磺酸;
步驟(2):向步驟(1)所得的預組裝溶液中依次加入乙二醇二甲基丙烯酸酯、偶氮二異丁腈、fe3o4@sio2@ch=ch2,通入氮氣30min,60℃機械攪拌24h,將攪拌所得物磁性分離,乙腈洗滌至上清液澄清後,甲醇/乙酸(v:v=9:1)混合溶液去除模板,甲醇洗滌至溶液呈中性,磁性分離,真空乾燥得到磁性分子印跡聚合物,其中,全氟辛烷磺酸、丙烯醯胺、乙二醇二甲基丙烯酸酯、偶氮二異丁腈和fe3o4@sio2@ch=ch2的質量比為1:14.22:79.3:4.50:10。
本發明中,以fe3o4@sio2@ch=ch2為核殼支撐結構、全氟辛烷磺酸做模板分子、乙二醇二甲基丙烯酸酯為交聯劑、偶氮二異丁腈為引發劑,採用本發明方法製備得到的磁性分子印跡聚合物,具有以下優勢:
(1)合成的磁性分子印跡聚合物粒徑小(58.4nm),比表面積大,能在表面提供更多的結合位點,從而增大吸附量,提高分離富集效率;
(2)在fe3o4@sio2表面形成的分子印跡薄膜厚度僅約3.7nm,分子印跡的結合位點大多位於印跡聚合物表面,吸附pfos的平衡時間較短,同時吸附效率高;
(3)磁性分子印跡聚合物吸附pfos達到平衡後,在洗脫劑的作用下能很好的洗脫模板,從而在重複吸附-脫附5次後,磁性分子印跡聚合物對pfos的吸附量基本保持不變,重複利用效果好;
(4)以模板為pfos,與單體丙烯醯形成的複合物在fe3o4@sio2@ch=ch2為支撐體、乙二醇二甲基丙烯酸酯為交聯劑的條件下,在洗去模板後能形成對pfos結構單一的空間網狀結構,對pfos的特異選擇性好。
附圖說明
圖1是磁性分子印跡聚合物的透射電鏡圖;
圖2是磁性分子印跡聚合物的紅外光譜圖;
圖3是磁性分子印跡聚合物的x-射線衍射圖;
圖4是磁性分子印跡聚合物的磁滯回線圖;
圖5a、圖5b、圖5c和圖5d分別是磁性分子印跡聚合物的等溫吸附曲線、溫度對靜態吸附的影響曲線、langmuir等溫吸附模型和freundlich等溫模型;
圖6是磁性分子印跡聚合物的吸附動力學圖;
圖7是磁性分子印跡聚合物的特異選擇性吸附圖;
圖8是磁性分子印跡聚合物的重複使用圖。
具體實施例
實施例1fe3o4@sio2@ch=ch2的製備
步驟a稱取2.000gfecl3·6h2o完全溶解於60.0ml乙二醇中,形成黃色的透明溶液,依次加入4.000g無水醋酸鈉和15.2ml的1,6-己二胺,50℃下機械攪拌30min,轉入高溫反應釜在198℃反應6h,磁性分離,用超純水和乙醇洗滌,真空乾燥製得fe3o4。
步驟b稱取450.0mg步驟a所得物分散在240ml乙醇和60ml水中,超聲15min,加入7.5mlnh3·h2o和1.05mlteos,40℃下機械攪拌12h,磁性分離,用超純水和乙醇洗滌,真空乾燥製得fe3o4@sio2。
步驟c稱取1.000g步驟b所得物分散在160ml10%的醋酸溶液中,加0.6ml3-(異丁烯醯氧)丙基三甲氧基矽烷(mps),60℃下機械攪拌5h,磁性分離,用超純水和乙醇洗滌,真空乾燥製得fe3o4@sio2@ch=ch2。
實施例2磁性分子印跡聚合物的製備
取100.0mg全氟辛烷磺酸、1.422g丙烯醯胺加入到300ml乙腈中,常溫下機械攪拌12h,形成預組裝溶液。在所述預組裝溶液中依次加入7.930g乙二醇二甲基丙烯酸酯、0.450g偶氮二異丁腈、1.000gfe3o4@sio2@ch=ch2,通氮氣30min,60℃機械攪拌24h。將合成的磁性分子印跡聚合物磁性分離,用乙腈洗滌至上清液澄清,而後甲醇/乙酸(v:v=9:1)去除模板,直至上清液中檢測不到全氟辛烷磺酸的存在,用甲醇洗至溶液呈中性,磁性分離,真空乾燥。
實施例3磁性分子印跡聚合物的製備
取100.0mg全氟辛烷磺酸,0.710g丙烯醯胺加入到300ml乙腈中,常溫下機械攪拌12h,形成預組裝溶液。在所述預組裝溶液中依次加入7.930g乙二醇二甲基丙烯酸酯、0.450g偶氮二異丁腈、1.000gfe3o4@sio2@ch=ch2,通氮氣30min,60℃機械攪拌24h。將合成的磁性分子印跡聚合物磁性分離,用乙腈洗滌至上清液澄清,而後甲醇/乙酸(v:v=9:1)去除模板,直至上清液中檢測不到全氟辛烷磺酸的存在,用甲醇洗至溶液呈中性,磁性分離,真空乾燥。
實施例4磁性分子印跡聚合物的製備
取100.0mg全氟辛烷磺酸,2.843g丙烯醯胺加入到300ml乙腈中,常溫下機械攪拌12h,形成預組裝溶液。在所述預組裝溶液中依次加入7.930g乙二醇二甲基丙烯酸酯、0.450g偶氮二異丁腈、2.000gfe3o4@sio2@ch=ch2,通氮氣30min,60℃機械攪拌24h。將合成的磁性分子印跡聚合物磁性分離,用乙腈洗滌至上清液澄清,而後甲醇/乙酸(v:v=9:1)去除模板,直至上清液中檢測不到全氟辛烷磺酸的存在,用甲醇洗至溶液呈中性,磁性分離,真空乾燥。
實施例5磁性分子印跡聚合物的透射電鏡表徵
實施例2製備的磁性分子印跡聚合物的透射電鏡圖見圖1,合成的磁性分子印跡聚合物具有較小的直徑,約58.4nm,由於合成的fe3o4@sio2的直徑約為51.0nm,故其分子印跡薄膜厚約3.7nm,超薄的分子印跡薄膜有助於該磁性分子印跡聚合物吸附全氟辛烷磺酸獲得較快的吸附動力平衡。
實施例6磁性分子印跡聚合物的紅外光譜表徵
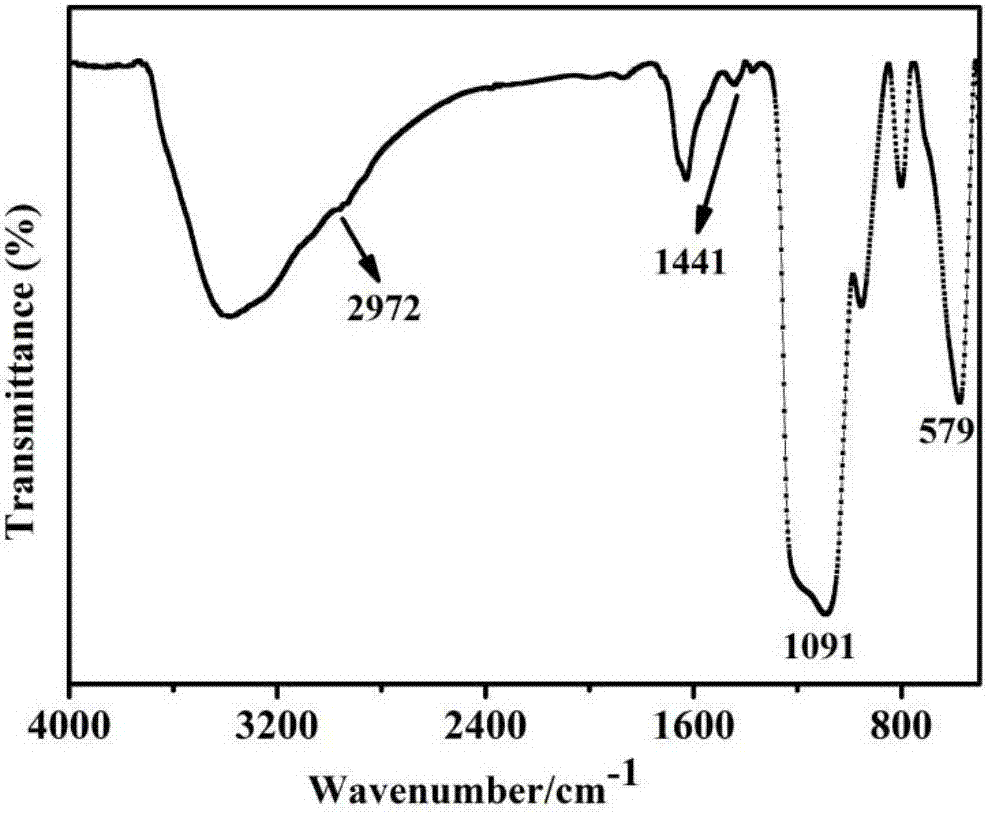
實施例2製備的磁性分子印跡聚合物的紅外光譜圖見圖2,579cm-1是fe-o的特徵吸收峰,表明四氧化三鐵成功地製備;1091cm-1是si–o–si的伸縮振動峰,表明二氧化矽成功地包被在四氧化三鐵的表面;1400cm-1是甲基的對稱彎曲振動,表明雙鍵功能化的四氧化三鐵成功製備;2972cm-1是乙二醇二甲基丙烯酸酯中的c-h伸縮振動,表明磁性分子印跡聚合物成功地合成。
實施例7磁性分子印跡聚合物的x--射線衍射分析
實施例2製備的磁性分子印跡聚合物的xrd圖見圖3,磁性分子印跡聚合物的6組特徵衍射峰(2θ=30.1°,35.5°,43.2°,53.6°,57.0°,和62.5°),及其峰位置(220),(311),(400),(422),(511),和(440)均能與標準磁性四氧化三鐵譜對應,表明四氧化三鐵呈立方尖晶石相,且合成過程中四氧化三鐵的晶體結構保持不變。
實施例8磁性分子印跡聚合物的磁性分析
實施例2製備的磁性分子印跡聚合物的磁性回線圖見圖4,準確地通過原點,且關於原點對稱,表明其剩磁和矯頑力均為零,說明該聚合物具有超順磁性;另外其飽和磁化強度為37.96emu/g,表明該聚合物在外加磁場的作用下,能快速分離富集水樣中的全氟辛烷磺酸。
實施例9磁性分子印跡聚合物的等溫吸附分析
5.0mg磁性分子印跡聚合物,200.0μl的br緩衝,1.0×10-4m的全氟辛烷磺酸100~700μl,稀釋至2.0ml。在培養搖床上25℃振搖60min,結果見圖5a,隨著pfos濃度的增大,磁性分子印跡聚合物(mmips)/非分子印跡聚合物(mnips)對pfos的吸附量均逐漸增大,當pfos的濃度為25.0μm時,吸附量最大,而後,隨著pfos的濃度的增加,吸附量不在變化,說明吸附達到平衡;同時mmips和mnips對pfos的吸附量差距比較大,也可以說明mmips對pfos的選擇性吸附。
實施例10溫度對磁性分子印跡聚合物吸附量的影響
5.0mg實施例2製備的磁性分子印跡聚合物,200.0μl的br緩衝,1.0×10-4m的全氟辛烷磺酸100~700μl,稀釋至2.0ml。在培養搖床上25℃、45℃和65℃,結果見圖5b-5d,隨著溫度升高,吸附量增大,說明該吸附過程屬於吸熱過程;並對實驗各個溫度下的吸附做langmuir和freundlich等溫吸附分析如圖5c和5d,公式分別為lnqe=lnkf+nlnce,發現該等溫吸附屬於langmuir等溫吸附模型,表現出單分子層吸附。
實施例11磁性分子印跡聚合物的吸附動力學分析
5.0mg實施例2製備的磁性分子印跡聚合物,200.0μl的br緩衝,1.0×10-4m的全氟辛烷磺酸500.0μl,稀釋至2.0ml。在培養搖床上25℃振搖不同時間(10min,20min,30min,40min,50min,60min,80min和100min),結果見圖6,隨著時間的延長,磁性分子印跡聚合物(mmips)/非分子印跡聚合物(mnips)對全氟辛烷磺酸的吸附量均逐漸增大,當時間為60min時,吸附達到平衡,故平衡吸附時間為60min。
實施例12磁性分子印跡聚合物的特異選擇性分析
5.0mg實施例2製備的磁性分子印跡聚合物,200.0μl的br緩衝,25.0μm的pfos與其中一個相同濃度的pfoa、sds、sdbs,稀釋至2.0ml。在25℃培養搖床上振搖60min,結果見圖7,當pfos的結構類似物存在時,磁性分子印跡聚合物對pfos的吸附量基本保持不變,說明合成的磁性分子印跡聚合物對pfos有特異選擇性吸附,另外由mmips和mnips對pfos吸附可得出同樣的結論。
實施例13磁性分子印跡聚合物的重複使用
磁性分子印跡聚合物的吸附-脫附實驗是在pfos的濃度為25.0μm,且以甲醇/乙酸(v:v=9:1)作為洗脫劑的條件下完成的,結果見圖8,在磁性分子印跡聚合物重複吸附-脫附5次後,其對pfos的吸附量基本保持不變,說明該磁性分子印跡聚合物具有較好的重複利用效果。