一種用BseNI鑑定人乳腺癌MALAT1基因rs619586多態性的方法與流程
2023-12-10 04:03:21 2
本發明屬於生物技術領域,涉及一種用BseNI鑑定人乳腺癌MALAT1基因rs619586多態性的方法。
背景技術:
單核苷酸多態性(single nucleotide polymorphism,SNP),主要是指在基因組水平上由單個核苷酸的變異所引起的DNA序列多態性,包括鹼基的置換/插入和缺失等形式。SNP是人類可遺傳的變異中最常見的一種,現已廣泛用於簡單和複雜疾病的遺傳連鎖分析/關聯分析及疾病易感基因的定位,指導易感基因克隆.較常見的單鹼基多態性位點的檢測方法包括聚合酶鏈式反應-限制性片段長度多態性技術(PCR-RFLP),三引物等位基因擴增法(TP-PCR)和測序等方法。這些方法雖各有優點,但也各有其不足之處。
PCR-RFLP是一種快速,簡便,準確的檢測SNP基因型的經典方法。其原理是:限制性內切酶是一類識別DNA特異位點(通常4-6bp),並在特異位點進行切割的酶類。酶切位點的特異性意味著對特定等位基因的完全消化會產生同樣的片段序列。而鹼基的置換,插入和缺失可以產生或消除一個特定酶切位點,從而改變酶切後產生片段的大小和數目。這些酶切片段帶型的不同稱為限制性片段長度多態性。如果SNP產生和消除了某個限制性內切酶位點,則可以通過對PCR產物進行酶切、電泳加以檢測。該方法的優點在於快速簡單,終點判斷準確。但其主要缺點在於酶切位點的選擇,要求待測多態性位點和某一酶切位點相關。PCR-RFLP酶的選擇具有局限性,並不是所有的SNP位點都可以用這種方法來鑑別,且部分內切酶價格昂貴,實驗成本高。三引物等位基因擴增法(TP-PCR)是一種不需要限制性內切酶和連接酶的重組DNA方法。反應體系中有2種模板和3種引物,從而在同一個反應體系中產生一個重組DNA分子。這種方法的主要優點是快速,不依賴連接片段的限制酶位點。但其缺點是擴增效率低,擴增特異性差,無法保證PCR產物的特異性和穩定性,分型結果易造成誤判。Taqman螢光探針法是一種新型高效準確的基因分型方法,其優點是檢測靈敏度高,分型準確。但該方法需要昂貴的儀器和較長的步驟,成本較高。
肺腺癌轉移相關轉錄子1(metastasis associated lung adenocarcinoma transcript 1,MALAT1)其編碼基因位於人類染色體11q13.1。MALAT 1長約8kb,為高度保守的長鏈非編碼RNA,在多種組織中廣泛地表達。在細胞水平,MALAT 1通過控制轉錄和重要細胞調控基因的mRNA前體加工過程來調節細胞增殖。MALAT1在包括乳腺癌在內的多種腫瘤組織中表達上調,且MALAT1過度表達可以通過下調腫瘤抑制因子PSF等方式,參與腫瘤細胞的生長、增殖、轉移以及免疫調節等環節,促進腫瘤的發生發展。另有研究表明,MALAT1是乳腺癌的危險因素,能夠使乳腺癌的發病風險增加0.97倍,若其發生基因突變將可能導致DNA損傷修復能力降低,長期積累的突變有可能促使細胞發生癌變以及腫瘤的發生和發展。
MALAT1基因rs619586位點多態性在人群中存在三種基因型:AA型(人基因組rs619586多態位點的兩個等位基因鹼基均為A),AG型(人基因組rs619586多態位點的兩個等位基因鹼基分別為A和G)和GG型(人基因組rs619586多態位點的兩個等位基因鹼基均為G)(美國國立衛生研究院國立醫學圖書館生物信息技術中心,http://www.ncbi.nlm.nih.gov)。
目前人MALAT1基因多態rs619586的鑑定常採用PCR-RFLP方法。但是目前鑑定人MALAT1基因多態rs619586時常使用的限制性內切酶價格昂貴(關於部分內切酶的參考價格可參考後面的表1),實驗成本高,難以在實驗室中普及使用。
技術實現要素:
為了克服現有技術中存在的缺陷,本發明提供一種操作簡單、成本低、適用範圍廣泛的用BseNI鑑定人乳腺癌MALAT1基因rs619586多態性的方法,通過PCR-RFLP技術的改進,來開發經濟、快速的檢測人MALAT1rs619586基因多態性的試劑盒。
其技術方案如下:
一種用BseNI鑑定人乳腺癌MALAT1基因rs619586多態性的方法,包括以下步驟:
(a)抽提樣品的基因組DNA;
(b)提供擴增人MALAT1基因多態性rs619586位點附近序列的正向引物和反向引物,以所述待測人基因組DNA為模板,進行PCR擴增反應,獲取擴增產物。
(c)使用限制性內切酶對所述擴增產物進行酶切,獲取相應的酶切產物;
(d)將酶切產物採用3%的瓊脂糖凝膠進行電泳,以判定MALAT1基因多態性rs619586位點的各基因型。
進一步,步驟(b)中所述正向引物和反向引物的設計與合成具體為:鹼基序列信息從NCBI的dbSNP資料庫獲取,在CHIP網站上進行核對,確定MALAT1多態位點鹼基變異數據;含MALAT1rs619586的部分基因序列如SEQ:ID:NO:1所示;基因序列的第501個鹼基R代表了A/G多態,即為rs619586的多態性位點。根據rs619586的具體序列和多態性鹼基的位置採用Primer Premier 6.0設計出的最有上下遊引物,正向引物序列如SEQ:ID:NO:2所示;
反向引物序列如SEQ:ID:NO:3所示,其中反向引物序列中倒數第4位鹼基為C,錯配後PCR產物中多態附近序列由ACTGTC或GCTGTC更改為ACTGGC或GCTGGC,擴增產物中A等位基因片段可被識別ACTGGN序列的限制性內切酶BseNI識別,根據片段切開情況來判斷多態基因型。按照正向引物序列和反向引物序列合成引物,引物的合成採用固相合成法,或委託生物公司合成並檢測正向和反向引物。
進一步,步驟(b)中PCR擴增反應中製備PCR擴增體系具體為:2×Taq PCR Mix7.5μl,雙蒸水5.9μl,上遊引物0.3μl下遊引物0.3μl以及模板DNA1.0μl,充分混勻後即得15.0μlPCR擴增反應體系;按照第一階段:94℃變性5min,第二階段共包括35個循環三個步驟,首先94℃變性30s,57.6℃退火45s,最後72℃延伸45s,第三階段:72℃延伸5min,最終4℃儲存以備用,即得長為154bp的PCR擴增產物。擴增後的產物序列如SEQ:ID:NO:4所示。
進一步,步驟(c)中進行酶切的酶切體系具體為:PCR反應產物5μl,限制性內切酶0.5μl,Buffer1.0μl以及無核酸酶純水8.5μl共計15μl酶切體系,於水浴鍋中37℃酶切1-16h,即得酶切產物,選擇BseNI進行酶切鑑定。
進一步,步驟(d)瓊脂糖凝膠電泳,確定基因多態性具體為:將酶切產物用3%的瓊脂糖凝膠在4-10V/cm的條件下,電泳20-40min,至紫外燈下能分清明顯的條帶後並拍照鑑定。對於不同的基因型,rs619586位點不同基因型展現出不同的條帶,野生基因型AA,長為134bp,20bp兩條帶;雜合基因型AG,長為154bp,134bp及20bp三條帶;雜合基因型GG,154bp一條帶。
與現有技術相比,本發明的有益效果:
本發明採用創造酶切位點法(Created Restriction Site PCR,CRS-PCR),利用引物鹼基錯配技術設計檢測MALAT1多態性rs619586。由於這種方法應用了引物3』端錯配技術,PCR產物進行酶切電泳後即可進行基因型的鑑定工作,因此在實際運用中具有很大的靈活性,且檢測方法簡單易行,是一種進行單鹼基突變位點基因型鑑定的較好方法。本發明建立的MALAT1多態性rs619586基因分型方法簡單、快速、安全、準確、靈敏度高,值得在臨床和研究中推廣,並可借鑑用於其它多態性基因的分型。
附圖說明
圖1是MALAT1rs619586位點的電泳圖譜;
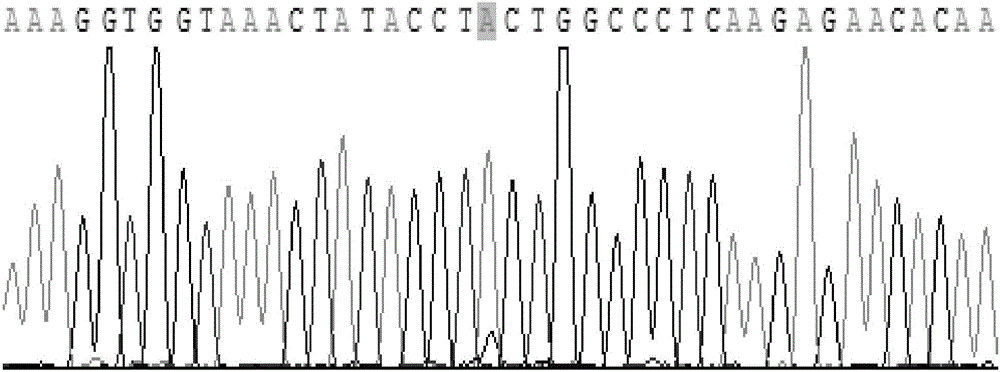
圖2是MALAT1rs619586位點AA基因型測序圖譜;
圖3是MALAT1rs619586位點AG基因型測序圖譜;
圖4是MALAT1rs619586位點GG基因型測序圖譜。
具體實施方式
下面結合附圖和實施例進一步說明本發明的技術方案。
本發明的方法包括以下步驟:
(a)抽提樣品的基因組DNA;
(b)提供擴增人MALAT1基因多態性rs619586位點附近序列的正向引物和反向引物,以所述待測人基因組DNA為模板,進行PCR擴增反應,獲取擴增產物。
(c)使用限制性內切酶對所述擴增產物進行酶切,獲取相應的酶切產物;
(d)將酶切產物採用3%的瓊脂糖凝膠進行電泳,以判定MALAT1基因多態性rs619586位點的各基因型。
其中,所述反向引物其3』末端倒數第1位鹼基與多態rs619586鹼基相鄰,倒數第4位鹼基為C(擴增產物的對應位置為錯配鹼基G),以便在擴增產物中與多態A等位基因形成ACTGGN結構(第1個鹼基A為多態等位基因,第5個鹼基G為錯配鹼基)。
本發明的方法可以通過引物上的錯配鹼基將SNP附近的序列改變為可被預先確定的限制性內切酶識別的序列。因此,可以克服傳統的PCR-RFLP方法所存在的需要採用昂貴內切酶的缺陷,進而大大降低了檢測SNP的成本。具體而言:由於該反向引物序列中倒數第4位鹼基為C(擴增產物的對應位置為錯配鹼基G),因此錯配後PCR產物中多態附近序列由ACTGTC或GCTGTC更改為ACTGGC或GCTGGC(其中第1個鹼基A/G為多態位點,第5個鹼基G為錯配鹼基)。因此擴增產物中A等位基因片段可被識別ACTGGN序列的限制性內切酶識別(即A等位基因片段可被內切酶切開);進而,可以根據片段切開情況來判斷多態基因型。更具體地,經序列分析可知,基因原始序列中A/G多態可由表1中前兩種內切酶識別。如通過鹼基錯配PCR將多態性位點後面第4位鹼基T改變為G,則該多態為A等位基因經PCR擴增後包含ACTGGC序列可被識別ACTGGN序列的限制性內切酶BseNI識別,而G等位基因不能切開。
經序列分析發現,檢測該A/G多態的方法可使用表1中的3種酶,其中表中前兩種酶價格昂貴,且酶切效果不理想。在採用創造酶切位點法(Created Restriction Site PCR,CRS-PCR)對反向引物序列中倒數第4位鹼基由A改變為C後,PCR產物中多態附近序列生成ACTGGC序列,從而可被限制性內切酶BseNI識別。由於識別特定位點的限制性內切酶BseNI適用範圍廣,價錢較為經濟,進而大大降低了檢測SNP的成本。因此,綜合考慮簡單操作性與經濟實用性,限制性內切酶BseNI是最好選擇。
表1中提供的限制性內切酶的價格從NEB公司(http://www.neb-china.com),限制性內切酶的選擇從WatCut限制性內切酶分析軟體上獲取(http://watcut.uwaterloo.ca/template.php?act=snp_new),以此作為限制性內切酶識別位點和價格的參考。
表1幾種限制性內切酶識別序列及其價格
在本發明的實施方案中,正向引物和反向引物的設計採用Primer6.0軟體進行,設計的原則綜合考慮引物對PCR擴增的靈敏度、特異度以及擴增效率的影響。按照鹼基互不配對的原則設計引物,引物長度一般在15~30鹼基之間,過長或過短均會造成特異性差,過長還會導致其延伸溫度大於74℃,不適於TaqDNA聚合酶進行反應。引物GC含量在40%~60%之間,Tm值最好接近72℃,GC含量過高或過低都不利於引發反應。鹼基要隨機分布,引物自身及引物之間不應存在互補序列,否則引物自身會摺疊成髮夾結構使引物本身復性。引物5′端和中間ΔG值應該相對較高,而3′端ΔG值較低。擴增產物的單鏈不能形成二級結構。引物應具有特異性,引物設計完成以後,應對其進行BLAST檢測,以確保其與其它基因不具有互補性。
最終正向引物由選自下列的核苷酸序列構成:GGGAGAAAGTCCGCCATTTTGCCAC。
反向引物由下列核苷酸序列構成:CTTCTTGTGTTCTCTTGAGGGCCAG。
正向引物和反向引物的設計主要考慮引物對PCR擴增的靈敏度、特異度和擴增效率的影響。通常按照鹼基互補配對原則設計引物,引物與模板的序列要緊密互補。引物長度為15~30bp,過短或過長可導致特異性差,過長亦會導致其延伸溫度大於74℃而不利於PCR反應。反向引物在其3,末端倒數第4位含有鹼基C,以便在擴增產物中與多態A等位基因形成ACTGGC結構,從而可以被BseNI內切酶識別。擴增產物總長度為154bp.擴增產物如下:GGGAGAAAGTCCGCCATT TTGCCACTTC TCAACCGTCC CTGCAAGGCT GGGGCTCAGT TGCGTAATGG AAAGTAAAGC CCTGAACTAT CACACTTTAA TCTTCCTTCA AAAGGTGGTA AACTATACCT R CTGGCCCTCA AGAGAACACA AGAAG
下劃線部分分別為正向引物和反向引物,第129位鹼基R代表A/G多態即SNP位點rs619586。第133位鹼基為錯配後的鹼基G(原為鹼基T)。該長為154bp的擴增產物經限制性內切酶BseNI酶切後可產生154bp,134bp,20bp共三種片段類型。基因型判定結果:野生基因型AA,長為134bp,20bp兩條帶;雜合基因型AG,長為154bp,134bp及20bp三條帶;雜合基因型GG,154bp一條帶。
凝膠電泳分為瓊脂糖凝膠電泳和聚丙烯醯胺凝膠電泳,瓊脂糖(Agarose)是一種線性多糖聚合物,系從紅色海藻產物瓊脂中提取而來的。當瓊脂糖溶液加熱到沸點後冷卻凝固便會形成良好的電泳介質,其密度是由瓊脂糖的濃度決定的,可用於DNA片段的製備電泳。聚丙烯醯胺凝膠主要有兩種方式:一是用於分離和純化雙鏈DNA片段的非變性聚丙烯醯胺凝膠,二是用於分離及純化單鏈DNA片段的變性聚丙烯醯胺凝膠。但綜合考慮實用性、便利性以及經濟性,使用瓊脂糖凝膠電泳不失為一種最佳選擇。在本發明中,目的擴增產物使用限制性核酸內切酶BseNI3U水浴鍋中酶切1-16h。酶切產物採用3%的瓊脂糖進行瓊脂糖凝膠電泳,在紫外燈照射下根據不同的條帶判斷野生純合基因型,雜合基因型以及突變純合基因型。
在本發明的具體實施方案中,MALAT1基因多態rs619586的具體步驟如下:
(l)提取待測人基因組DNA模板,所述人基因組DNA模板為人體任何部分取得的人基因組模板。
(2)PCR擴增基因組DNA,對提取的模板基因組DNA進行PCR擴增,獲得含多態附近序列的PCR產物。
(3)對PCR擴增產物用限制性核酸內切酶BseNI進行酶切反應,得到酶切產物;
(4)酶切產物進行瓊脂糖進行瓊脂糖凝膠電泳,在紫外燈照射下根據不同的條帶判斷野生純合基因型,雜合基因型以及突變純合基因型。
下面對上述各個主要步驟進行詳細描述:
(1)引物設計與合成
鹼基序列信息從NCBI的dbSNP資料庫獲取,在CHIP網站上進行核對,確定MALAT1多態位點鹼基變異數據.
含MALAT1rs619586的部分基因序列如下:
GGAGACAAAG CCATTCGCTT AGTTGGTCTA CTTTAAAAGG CCACTTGAAC TCGCTTTCCA
TGGCGATTTG CCTTGTGAGC ACTTTCAGGA GAGCCTGGAA GCTGAAAAAC GGTAGAAAAA
TTTCCGTGCG GGCCGTGGGG GGCTGGCGGC AACTGGGGGG CCGCAGATCA GAGTGGGCCA
CTGGCAGCCA ACGGCCCCCG GGGCTCAGGC GGGGAGCAGC TCTGTGGTGT GGGATTGAGG
CGTTTTCCAA GAGTGGGTTT TCACGTTTCT AAGATTTCCC AAGCAGACAG CCCGTGCTGC
TCCGATTTCT CGAACAAAAA AGCAAAACGT GTGGCTGTCT TGGGAGCAAG TCGCAGGACT
GCAAGCAGTT GGGGGAGAAA GTCCGCCATT TTGCCACTTC TCAACCGTCC CTGCAAGGCT
GGGGCTCAGT TGCGTAATGG AAAGTAAAGC CCTGAACTAT CACACTTTAA TCTTCCTTCA
AAAGGTGGTA AACTATACCT
A CTGTCCCTCA AGAGAACACA AGAAGTGCTT TAAGAGGTAT TTTAAAAGTT CCGGGGGTTT
TGTGAGGTGT TTGATGACCC GTTTAAAATA TGATTTCCAT GTTTCTTTTG TCTAAAGTTT
GCAGCTCAAA TCTTTCCACA CGCTAGTAAT TTAAGTATTT CTGCATGTGT AGTTTGCATT
CAAGTTCCAT AAGCTGTTAA GAAAAATCTA GAAAAGTAAA ACTAGAACCT ATTTTTAACC
GAAGAACTAC TTTTTGCCTC CCTCACAAAG GCGGCGGAAG GTGATCGAAT TCCGGTGATG
CGAGTTGTTC TCCGTCTATA AATACGCCTC GCCCGAGCTG TGCGGTAGGC ATTGAGGCAG
CCAGCGCAGG GGCTTCTGCT GAGGGGGCAG GCGGAGCTTG AGGAAACCGC AGATAAGTTT
TTTTCTCTTT GAAAGATAGA GATTAATACA ACTACTTAAA AAATATAGTC AATAGGTTAC
TAAGATATTG CTTAGCGTTA
基因序列的第501個鹼基R代表了A/G多態,即為rs619586的多態性位點。根據rs619586的具體序列和多態性鹼基的位置採用Primer Premier 6.0設計出的最有上下遊引物如下;
正向引物序列:5』-GGGAGAAAGTCCGCCATTTTGCCAC-3』
反向引物序列:5』-CTTCTTGTGTTCTCTTGAGGGCCAG-3』。
其中反向引物序列中倒數第4位鹼基為C(形成的擴增產物對應位置為錯配鹼基G),因此錯配後PCR產物中多態附近序列由ACTGTC或GCTGTC更改為ACTGGC或GCTGGC(其中第1個鹼基A/G為多態位點,第5個鹼基G為錯配鹼基)。因此擴增產物中A等位基因片段可被識別ACTGGN序列的限制性內切酶BseNI識別(即A等位基因片段可被內切酶切開)。進而,可以根據片段切開情況來判斷多態基因型。
按照正向引物序列和反向引物序列合成引物,引物的合成可以採用本領域通用的方法(如固相合成法),亦可委託生物公司合成並檢測正向和反向引物。
(2)製備PCR擴增體系:2×Taq PCR Mix7.5μl,雙蒸水5.9μl,上遊引物0.3μl下遊引物0.3μl以及模板DNA1.0μl,充分混勻後即得15.0μl PCR擴增反應體系;按照第一階段:94℃變性5min,第二階段共包括35個循環三個步驟,首先94℃變性30s,57.6℃退火45s,最後72℃延伸45s,第三階段:72℃延伸5min,最終4℃儲存以備用,即得長為154bp的PCR擴增產物。擴增後的產物序列為:
GGGAGAAA GTCCGCCATT TTGCCACTTC TCAACCGTCC CTGCAAGGCT
GGGGCTCAGT TGCGTAATGG AAAGTAAAGC CCTGAACTAT CACACTTTAA TCTTCCTTCA
AAAGGTGGTA AACTATACCT R CTGGCCCTCA AGAGAACACA AGAAG
下劃線部分分別為正向引物和反向引物,第129位鹼基R代表A/G多態即SNP位點rs619586。第133位鹼基為錯配後的鹼基G(原為鹼基T)。
(3)酶切體系:PCR反應產物5μl,限制性內切酶0.5μl,Buffer1.0μl以及無核酸酶純水8.5μl共計15μl酶切體系,於水浴鍋中37℃酶切1-16h,即得酶切產物。綜合考慮經濟實用性以及簡單操作性最終選擇BseNI進行酶切鑑定。
(4)瓊脂糖凝膠電泳,確定基因多態性:將酶切產物用3%的瓊脂糖凝膠在4-10V/cm的條件下,電泳20-40min,至紫外燈下能分清明顯的條帶後並拍照鑑定。對於不同的基因型,rs619586位點不同基因型展現出不同的條帶(見圖1),野生基因型AA,長為134bp,20bp兩條帶;雜合基因型AG,長為154bp,134bp及20bp三條帶;雜合基因型GG,154bp一條帶。各基因型均經測序以進一步鑑定,測序結果(見圖2-4)顯示與現有技術測得的結果完全相同。
實施例1.人乳腺癌外周血全血標本測定人MALAT1rs619586多態性
1材料與方法
1.1主要儀器與試劑
儀器:BCD-228CH冰箱(新飛電器),HH-2數顯恆溫水浴鍋(華峰儀器),SmartGe凝膠成像儀(北京賽智創業),GT9612梯度PCR儀(百泰克生物),WD900SL23-2型號微波爐(格蘭仕電器),DG-300C型電泳儀(鼎國昌盛生物)等。
試劑:NEP004-1DNA提取試劑盒(鼎國昌盛生物),50bp DNA Ladder(萊風生物),2×Taq PCR Mix(萊風生物),BseNI(Thermo),瓊脂糖(SIGMA)等。
1.2引物設計
在NCBI的dbSNP資料庫中,查找MALAT1rs619586的核苷酸序列,在引物設計軟體Primer Premier 6.0中,設置引物長度和擴增目的片段長度等參數進行引物設計,根據GC含量和退火溫度等選擇最優上下遊引物,並將反向引物序列中倒數第4位鹼基設為C(形成的擴增產物對應位置為錯配鹼基G),因此錯配後PCR產物中多態附近序列由ACTGTC或GCTGTC更改為ACTGGC或GCTGGC(其中第二個鹼基A/G為多態位點,第5個鹼基G為錯配鹼基)。因此擴增產物中A等位基因片段可被識別ACTGGN序列的限制性內切酶BseNI識別(即A等位基因片段可被內切酶切開)。再將此引物與Pubmed中的Blast序列比對功能(http://blast.ncbi.nlm.nih.gov/Blast.cgi)進行引物比對,最終確定的上下遊引物為正向引物序列:
5』-GGGAGAAAGTCCGCCATTTTGCCAC-3』
反向引物序列:5』-CTTCTTGTGTTCTCTTGAGGGCCAG-3』。
1.3限制性內切酶的選擇
根據dbSNP中rs619586前後五個位點的鹼基序列,用WatCut在線限制性內切酶分析軟體,在線搜索獲得可識別突變位點的限制性內切酶的信息,綜合考慮其酶切特異性及經濟適用性選擇最佳的限制性內切酶BseNI。
1.4從人乳腺癌全血中提取待測樣本的基因組DNA
嚴格按照離心柱型DNA提取試劑盒操作步驟提取基因組DNA。
l)在1.5ml離心管中加入300μl血細胞後,再加入900μl細胞裂解液混和均勻,在冰上放置10min後,在離心機中12000rpm離心1min,棄上清,再次加入900μl細胞裂解液,用槍吹起沉澱並混勻後,重複上述步驟;
2)向沉澱物中加入600μl solution B溶液,用移液槍輕輕吹起沉澱後,加入10μl蛋白酶K混勻,在70℃水浴鍋中放置10min後,12000rpm離心5min。
3)將離心管中的上清轉入編過號的新的離心管中,再向新的離心管中加入500μl無水乙醇,期間可能會出現絮狀沉澱物,該絮狀物即為DNA;
4)將混勻的液體轉入離心柱中(若一次轉不完,可分次轉入),室溫靜置2min,再12000rpm離心1min,棄廢液;
5)加入700μl已加入相應體積無水乙醇的solution C漂洗液,室溫靜置2min,12000rpm離心1min,棄廢液;
6)加入700μl已加入相應體積無水乙醇的solution D漂洗液,室溫靜置2min,12000rpm離心1min,棄廢液;
7)加入500μl solution D漂洗液,12000rpm離心1min,棄廢液;
8)再次離心2min,將離心柱置於新的編過號的離心管中,敞口放入37℃恆溫箱內10min直至無明顯乙醇味;
9)在矽基質膜中央加入已經預熱到65℃的solution E 100μl,室溫放置5min,12000rpm離心1min,重複上述步驟一次,終離心後的液體即為提取出的基因組DNA;
10)吸取2μl提取出的DNA用NanoPhotometer Pearl微量分光光度計測定其濃度和純度。
2結果
2.1PCR擴增
(1)根據提取基因組DNA的濃度,將研究對象的DNA進行稀釋,使終濃度為20μg/μl。
(2)PCR擴增體系為2×Taq PCR Mix 7.5μl、上遊引物和下遊引物各0.3μl、模板DNA 1.0μl,最後用雙蒸水補充總體積至15μl。
(3)PCR反應條件:第一個階段為預變性階段,94℃/5min;第二個階段包括三個步驟共35個循環,依次設置為94℃/35s、58℃退火時間45s、72℃/30s;第三個階段72℃/5min。2.2酶切反應
酶切體系為PCR擴增產物5μl、限制性內切酶0.5μl、Buffer 1.0μl,最後用雙蒸水補充總體積至15μl。混勻後置於水浴鍋中37℃水浴1-16小時。
2.3基因型的判定
表2 rs619586位點基因型判定
實施例2.人乳腺癌組織標本測定人MALAT1rs619586多態性
與實施例1的步驟基本相同,只是採用下面的方法從乳腺癌組織標本中提取DNA作為待測DNA。
使用手機切除乳腺癌組織,酚-氯仿法提取乳腺癌組織基因組DNA作為待測DNA。
1)將乳腺癌組織塊解凍,用生理鹽水洗去血汙,剪取0.1g左右的組織碾磨,加入1ml的滅菌水,顛倒混勻,10000轉離心10分鐘,棄上清。管底應該有沉澱。重複兩次
2)加入200ul的DNA裂解液,5ul的蛋白酶K混勻,55度過夜消化。
3)消化完成後加入等體積的酚氯仿混合液(1:1),劇烈震蕩,使其變成奶咖色。12000轉離心10分鐘。
4)取上清,注意不要洗到中間介質以及下層液體。加入等體積的氯仿,顛倒混勻。12000轉離心10分鐘。
5)取上清,注意不要洗到中間介質以及下層液體。加入十分之一的醋酸鈉以及2.5倍的無水乙醇,顛倒混勻。
6)12000轉離心10分鐘。棄上清。
7)加入1ml 70%乙醇,使其白色沉澱懸浮,顛倒混勻數次。12000轉離心10分鐘。
8)棄上清,然後室溫開蓋靜置分鐘使其乙醇揮發乾淨。
9)加入滅菌水溶解DNA,一般加入50ul滅菌水溶解
10)-20℃保存,即得乳腺癌組織基因組DNA
結果:將酶切產物用3%的瓊脂糖凝膠電泳20-40min,至紫外燈下能分清明顯的條帶並拍照鑑定。對於不同的基因型,rs619586位點不同基因型展現出不同的條帶(見圖1),AA野生基因型,長為134bp,20bp兩條帶;AG雜合基因型,長為154bp,134bp及20bp三條帶;GG雜合基因型,154bp一條帶。各基因型均經測序以進一步鑑定,測序結果(見圖2-4)顯示與現有技術測得的結果完全相同。
從上述實施例可以看出,本發明針對使用CRS-PCR鑑定MALAT1rs619586多態性,對傳統PCR-RFLP法進行改進,應用了引物3』端錯配技術,克服了傳統PCR-RFLP法內切酶選擇餘地小,價格昂貴,實驗成本高等缺點,本研究建立的MALAT1rs619586多態性的檢測方法,內切酶價格十分便宜,同時PCR產物純度較高,酶切結果易於分辨。該檢測方法通過測序驗證,其結果準確可靠。
SNP檢測技術已經很成熟,較常見的單鹼基多態性位點的檢測方法包括測序、三引物等位基因擴增法(TP-PCR)、聚合酶鏈式反應-限制性片段長度多態性技術(PCR-RFLP)等方法。這些方法雖各有優點,但也各有其不足之處。測序可以直接測出DNA序列中的所有突變位點的鹼基替代情況,但該方法需要昂貴的儀器和較長的步驟,成本較高;TP-PCR方法準確性低,結果易造成誤判;PCR-RFLP方法比較成熟,但要求待測多態性位點和某一酶切位點相關,酶的選擇具有局限性,往往成本較高。
因此,鑑於本發明所使用的限制性內切酶價格低廉,且引物合成以及各試劑均較為經濟,加之CRS-PCR操作簡單,重複性好,因此是最經濟有效的基因型檢測方法。
本發明的對傳統PCR-RFLP技術進行改進,採用創造酶切位點法(Created Restriction Site PCR,CRS-PCR),利用引物鹼基錯配技術設計檢測MALAT1多態性rs619586。由於這種方法應用了引物3』端錯配技術,PCR產物進行酶切電泳後即可進行基因型的鑑定工作,因此在實際運用中具有很大的靈活性,且檢測方法簡單易行,是一種進行單鹼基突變位點基因型鑑定的較好方法。本項發明的技術關鍵點是設計出的MALAT1多態性rs619586的上下遊引物,正向引物序列:5』-GGGAGAAAGTCCGCCATTTTGCCAC-3』、
反向引物序列:5』-CTTCTTGTGTTCTCTTGAGGGCCAG-3』以及限制性內切酶BseNI的使用。總之,我們建立的MALAT1多態性rs619586基因分型方法簡單、快速、安全、準確、靈敏度高,值得在臨床和研究中推廣,並可借鑑用於其它多態性基因的分型。
以上所述僅為本發明最佳實施方式,任何熟悉本技術領域的技術人員在本發明披露的技術範圍內,可顯而易見地得到的技術方案的簡單變化或等效替換均落入本發明的保護範圍內。