芳胺氮芥衍生物N,N‑二(2‑氯乙基)‑N′‑丙醯基‑1,4‑苯二胺及其製備方法與流程
2023-11-01 03:40:57 6
本發明涉及氮芥類抗腫瘤藥物的技術領域,具體涉及芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺及其製備方法。
背景技術:
氮芥類藥物是常用的一種抗腫瘤藥物,具有特別好的抗腫瘤活性,但是作為傳統的細胞毒類藥物,其仍具有生物烷化劑所特有的缺點,在抑制和毒害增生活躍的腫瘤細胞的同時,對其它增生較快的正常細胞也產生抑制作用,例如對一些骨髓細胞、腸上皮細胞、還有一些毛髮細胞也具有阻擾滅殺的效果,這就會引起例如噁心、嘔吐、脫髮等嚴重的不良反應;另外,氮芥類藥物還具有免疫抑制作用,會降低機體的防禦功能,使細菌病毒等趁虛而入,引起各種感染。
氮芥類抗腫瘤藥物從構造上看由兩部分組成:烷基化部分和載體部分,烷基化部分是主體部分,也是抗腫瘤活性的功能基團,載體部分可看成結構修飾的部分,對其進行結構修飾可以不斷提高藥物在人體內的吸收、分布,進而有助於提高對腫瘤細胞的選擇性,同時也能有效的抑制副作用的產生。
因此,如何對氮芥衍生物進行修飾,合成出具有較低的毒副作用的氮芥,已成為目前研究的熱點。
技術實現要素:
針對現有技術中存在的問題,本發明的目的在於提供一種具有較低毒副作用的且能增強腫瘤治療效果,同時兼具殺菌消炎療效的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺,其可達到降低病人化療後因免疫能力下降而引起併發症的風險。
本發明的另一目的為提供上述芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的製備方法。
為了達到上述目的,本發明採用以下技術方案予以實現。
芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺,其特徵在於,結構如式Ⅰ:
上述芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的製備方法,其特徵在於,包括以下步驟:
步驟1,製備N,N-二(2-氯乙基)-1,4-苯二胺;
步驟2,向反應器中加入反應原料N,N-二(2-氯乙基)-1,4-苯二胺、二氯甲烷和三乙胺,冰水浴冷卻,攪拌,並向反應器中滴加丙醯氯和二氯甲烷的混合溶液,滴加完成後,撤掉冰水浴,在室溫下反應4-14小時,待反應完全後,使用蒸餾水洗滌反應液,且用無水硫酸銅乾燥,常壓蒸餾,對蒸餾後的濾餅進行純化,即得。
步驟1包含以下子步驟:
子步驟1a,首先按照以下步驟製備N,N-二(2-羥乙基)-4-硝基苯胺:將4-氯硝基苯、二乙醇胺、碳酸鉀和質量百分比為1%的CuSO4溶液依次加入反應器中,攪拌均勻,升溫至82℃時,向反應器中加入甲苯,繼續升溫至115-120℃,恆溫攪拌反應6-9小時,待反應完全後,蒸餾去除甲苯,將蒸餾後的母液倒入40℃-60℃的熱水中,攪拌,靜置過夜後抽濾水層得粗品,使用無水乙醇對所述粗品重結晶,得黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺。
優選地,子步驟1a中,所述4-氯硝基苯、二乙醇胺、碳酸鉀的摩爾比為(1.00∶1.20∶0.60)-(1.00∶1.40∶0.7)。
優選地,子步驟1a中,所述質量百分比為1%的CuSO4溶液與所述4-氯硝基苯的用量的比例為(0.2-0.5)mL:(2-3)g。
優選地,子步驟1a中,所述甲苯與所述4-氯硝基苯的用量的比例為1mL:(2-3)g。
子步驟1b,然後按照以下步驟製備N,N-二(2-氯乙基)-4-硝基苯胺:取所述黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺溶於裝有乾燥的二氯甲烷的反應器中,並向所述反應器中加入三乙胺;再在室溫下攪拌滴加二氯亞碸和二氯甲烷的混合溶液,所述滴加時間為1-1.5小時,升高溫度至40-45℃,回流,同時攪拌反應2小時,反應完全後靜置冷卻,並用飽和碳酸氫鈉溶液洗滌,合併有機層並用無水硫酸鈉乾燥,減壓蒸餾去除溶劑,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品,使用無水乙醇對所述粗品重結晶,得黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺。
優選地,子步驟1b中,所述黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺與所述二氯亞碸的摩爾比為1:1-1:2。
優選地,子步驟1b中,所述黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺與所述二氯甲烷、所述三乙胺的用量比例為2.7g:60mL:1.1mL。
優選地,子步驟1b中,所述二氯亞碸與所述二氯甲烷的體積比為1:(13-15)。
子步驟1c,最後按照以下步驟製備N,N-二(2-氯乙基)-1,4-苯二胺:取所述黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺溶於裝有濃鹽酸的反應器中,並向所述反應器中加入氯化亞錫,在氮氣氛圍下回流攪拌反應,待顏色變為無色透明時停止反應;使用0℃的冰水浴冷卻,並用濃氨水調pH至7.5~8.0,用乙酸乙酯萃取,無水硫酸鈉乾燥,減壓蒸餾溶劑,即得。
優選地,子步驟1c中,所述黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺與所述氯化亞錫、所述濃鹽酸的用量比為1mmol:(1-6)mol:(10-50)mL。
優選地,步驟2中,所述反應原料中,所述N,N-二(2-氯乙基)-1,4-苯二胺與所述二氯甲烷的用量比為1.0g:20.0mL;所述N,N-二(2-氯乙基)-1,4-苯二胺與所述三乙胺的摩爾比為1:3-1:5。
優選地,步驟2中,所述N,N-二(2-氯乙基)-1,4-苯二胺與所述丙醯氯的摩爾比為1:1.5-1:2.5;所述乙醯氯與所述二氯甲烷的混合溶液中,所述丙醯氯與所述二氯甲烷的摩爾比為1:4-1:6。
優選地,步驟2中,所述滴加的時間為1-2小時。
優選地,步驟2中,所述純化採用柱層析技術,所述柱層析技術中的流動相為石油醚與乙酸乙酯的混合物,且所述石油醚與所述乙酸乙酯的體積比為1:1-1:3。
與現有技術相比,本發明的有益效果為:
本發明以N,N-二(2-氯乙基)-1,4-苯二胺為藥效基團,以丙醯氯對N,N-二(2-氯乙基)-1,4-苯二胺進行結構修飾,使得本發明的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺在具有增強氮芥治療指數的前提下,又可以有效降低氮芥的毒副作用,同時兼具殺菌、消炎的療效,以達到降低病人化療後因免疫力下降而引起的併發症的風險。
附圖說明
下面結合附圖和具體實施例對本發明做進一步詳細說明。
圖1為N,N-二(2-羥乙基)-4-硝基苯胺的紅外光譜圖;圖中,橫坐標為波數,單位為cm-1,縱坐標為透光率,單位為%;
圖2為N,N-二(2-氯乙基)-4-硝基苯胺的紅外光譜圖;圖中,橫坐標為波數,單位為cm-1,縱坐標為透光率,單位為%;
圖3為N,N-二(2-氯乙基)-1,4-苯二胺的紅外光譜圖;圖中,橫坐標為波數,單位為cm-1,縱坐標為透光率,單位為%;
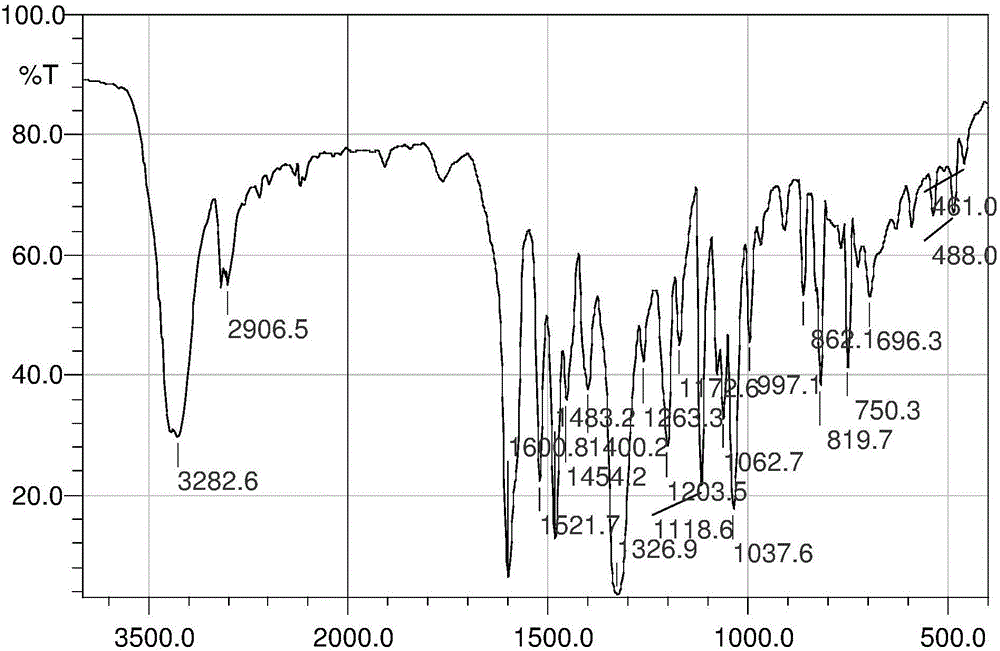
圖4為式Ⅰ的芳氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的紅外光譜圖;圖中,橫坐標為波數,單位為cm-1,縱坐標為透光率,單位為%;
圖5為式Ⅰ的芳氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的核磁譜圖;圖中,橫坐標是化學位移,單位為ppm。
具體實施方式
下面將結合實施例對本發明的實施方案進行詳細描述,但是本領域的技術人員將會理解,下列實施例僅用於說明本發明,而不應視為限制本發明的範圍。
本發明提供一種具有較低毒副作用的且能增強腫瘤治療效果,同時兼具殺菌消炎療效的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺,其結構如式Ⅰ:
上述芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的製備方法,包括以下步驟:
步驟1,製備N,N-二(2-氯乙基)-1,4-苯二胺;
子步驟1a,首先按照以下步驟製備N,N-二(2-羥乙基)-4-硝基苯胺:將4-氯硝基苯、二乙醇胺、碳酸鉀和質量百分比為1%的CuSO4溶液依次加入反應器中,攪拌均勻,升溫至82℃時,向反應器中加入甲苯,繼續升溫至115-120℃,恆溫攪拌反應6-9小時,待反應完全後,蒸餾去除甲苯,將蒸餾後的母液倒入40℃-60℃的熱水中,攪拌,靜置過夜後抽濾水層得粗品,使用無水乙醇對所述粗品重結晶,得黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺。
子步驟1b,然後按照以下步驟製備N,N-二(2-氯乙基)-4-硝基苯胺:取所述黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺溶於裝有乾燥的二氯甲烷的反應器中,並向所述反應器中加入三乙胺;再在室溫下攪拌滴加二氯亞碸和二氯甲烷的混合溶液,所述滴加時間為1-1.5小時,升高溫度至40-45℃,回流,同時攪拌反應2小時,反應完全後靜置冷卻,並用飽和碳酸氫鈉溶液洗滌,合併有機層並用無水硫酸鈉乾燥,減壓蒸餾去除溶劑,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品,使用無水乙醇對所述粗品重結晶,得黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺。
子步驟1c,最後按照以下步驟製備N,N-二(2-氯乙基)-1,4-苯二胺:取所述黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺溶於裝有濃鹽酸的反應器中,並向所述反應器中加入氯化亞錫,在氮氣氛圍下回流攪拌反應,待顏色變為無色透明時停止反應;使用0℃的冰水浴冷卻,並用濃氨水調pH至7.5~8.0,用乙酸乙酯萃取,無水硫酸鈉乾燥,減壓蒸餾溶劑,即得。
步驟2,向反應器中加入反應原料N,N-二(2-氯乙基)-1,4-苯二胺、二氯甲烷和三乙胺,冰水浴冷卻,攪拌,並向反應器中滴加丙醯氯和二氯甲烷的混合溶液,滴加完成後,撤掉冰水浴,在室溫下反應4-14小時,待反應完全後,使用蒸餾水洗滌反應液,且用無水硫酸銅乾燥,常壓蒸餾,對蒸餾後的濾餅進行純化,即得。
實施例1
1.1 N,N-二(2-羥乙基)-4-硝基苯胺的合成
向250mL三口燒瓶內分別加入4-氯硝基苯12.64g(0.080mol)、二乙醇胺10.99g(0.104mol)和7.18g(0.052mol)碳酸鉀,最後加入1.2mL質量百分比濃度為1%的CuSO4混合均勻。加熱升高溫度,當溫度達到82℃時,向反應液中加入5.0mL的甲苯,繼續升高溫度控制反應溫度為117℃,使用薄層色譜法跟蹤反應進程,恆溫攪拌反應6h。待反應完全後,蒸餾去除甲苯,將蒸餾後的母液倒入60℃熱水中,劇烈攪拌,靜置過夜後抽濾除去水層得粗品,粗品用無水乙醇重結晶,得黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺11.39g,收率63.0%。
1.2 N,N-二(2-氯乙基)-4-硝基苯胺的合成
稱取2.70g(0.012mol)黃色針狀晶體N,N-二(2-羥基乙基)-4-硝基苯胺,溶於60.0mL乾燥的二氯甲烷中,然後向反應瓶中加入1.1mL的三乙胺,取1.3ml(0.018mol)氯化亞碸,溶於20.0mL二氯甲烷,室溫攪拌下滴加該溶液於反應瓶,滴加時間為lh,滴加完成後,升溫至回流,在回流溫度下攪拌反應2h。反應完全後靜置冷卻,用30.0mL乾燥的二氯甲烷稀釋,分別用20.0mL的飽和碳酸氫鈉溶液洗滌三次,合併有機層用無水硫酸鈉乾燥,0.01MPa下減壓蒸餾除去溶劑二氯甲烷,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品。粗品用無水乙醇重結晶得黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺2.99g,收率95.0%。
1.3 N,N-二(2-氯乙基)-1,4-苯二胺的合成
稱取1.04g(4mmol)黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺溶於40.0mL濃鹽酸中,然後加入8.30g(20mmol)氯化亞錫,氮氣保護下回流待溶液顏色變為無色透明時停止反應。用0℃的冰水浴冷卻,加入20.0mL的水稀釋,用濃氨水調pH至7.5~8.0,用乙酸乙酯萃取三次,無水硫酸鈉乾燥,減壓蒸除溶劑,即得。由於新生成的化合物具有化學活性比較高的基團氨基,其化學結構不穩定易被氧化,因此在用乙酸乙酯萃取後,向其溶液中加入過量的濃鹽酸,製備其鹽酸鹽低溫避光備用,待應用時加鹼萃取即可使用,收率93.0%。
1.4式Ⅰ的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的合成
首先合成丙醯氯:在裝有回流冷凝管的250mL的三口燒瓶中加入丙酸,再加入丙酸質量1.5%的二甲基甲醯胺,在回流冷凝管的一段插入尾氣吸收裝置,攪拌、升溫至70-75℃,再緩慢滴加氯化亞碸,丙酸與氯化亞碸的摩爾比為1:1.5。待反應完成後,減壓除去過量的氯化亞碸及副產物,即得丙醯氯,對所製備的丙醯氯進行檢測,得無色的丙醯氯的純度大於98%,收率為97.0%,沸點為79℃,密度為1.06g/mL。
在裝有回流冷凝管的250mL的三口燒瓶中加入1.00g N,N-二(2-氯乙基)-1,4-苯二胺、20.0mL乾燥的二氯甲烷和1.2mL的三乙胺,冰水浴冷卻,攪拌下滴加丙醯氯和二氯甲烷的混合溶液15.0mL,滴加時間約為1.5小時,其中,N,N-二(2-氯乙基)-1,4-苯二胺與丙醯氯的物料配比為1:1.5,滴加結束後撤掉冰水浴,常溫反應6小時,反應結束後使用30mL的水洗滌反應液,無水硫酸鈉乾燥,常壓蒸餾,濾餅用柱層析(V(石油醚:乙酸乙酯)1∶1)純化得暗紅色晶體,即為最終產物,產物收率為91.1%。
實施例2
1.1 N,N-二(2-羥乙基)-4-硝基苯胺的合成
向250mL三口燒瓶內分別加入摩爾比為1:1.2:0.6的4-氯硝基苯、二乙醇胺和碳酸鉀,最後加入質量濃度為1%的CuSO4溶液,且質量濃度為1%的CuSO4溶液與4-氯硝基苯的用量比為0.5mL:3g,混合均勻;加熱升高溫度,當溫度達到82℃時,向反應液中加入甲苯,且甲苯與4-氯硝基苯的用量比為1mL:2g;繼續升高溫度控制反應溫度為120℃,使用薄層色譜法跟蹤反應進程,恆溫攪拌反應9h。待反應完全後,蒸餾去除甲苯,將蒸餾後的母液倒入60℃的熱水中,劇烈攪拌,靜置過夜後抽濾除去水層得粗品,粗品用無水乙醇重結晶,得黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺。
1.2 N,N-二(2-氯乙基)-4-硝基苯胺的合成
稱取2.70g(0.012mol)黃色針狀晶體N,N-二(2-羥基乙基)-4-硝基苯胺,溶於60.0mL乾燥的二氯甲烷中,然後向反應瓶中加入1.1mL的三乙胺;取1.3mL(0.018mol)二氯亞碸溶於20.0mL二氯甲烷溶液,室溫攪拌下將該溶液滴加到反應瓶,滴加的時間為l.5h,滴加完成後,加熱至45℃,回流,並在回流的狀態下攪拌反應2h。反應完全後靜置冷卻,用30.0mL乾燥的二氯甲烷稀釋,並分別用20.0mL的飽和碳酸氫鈉溶液洗滌三次,合併有機層用無水硫酸鈉乾燥,減壓蒸餾除去溶劑,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品。粗品用無水乙醇重結晶得黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺2.99g,收率95.0%。
1.3 N,N-二(2-氯乙基)-1,4-苯二胺的合成
稱取4mmol黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺溶於120.0mL濃鹽酸中,然後加入24mmol氯化亞錫,氮氣保護下回流待溶液顏色變為無色透明時停止反應。用0℃的冰水浴冷卻,加入20.0mL的水稀釋,用濃氨水調pH至7.5~8.0,用乙酸乙酯萃取3次,無水硫酸鈉乾燥,減壓蒸除溶劑,即得。由於新生成的化合物具有化學活性比較高的基團氨基,其化學結構不穩定易被氧化,因此在用乙酸乙酯萃取後,向其溶液中加入過量的濃鹽酸,製備其鹽酸鹽低溫避光備用,待應用時加鹼萃取即可使用。
1.4式Ⅰ的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的合成
首先合成丙醯氯:在裝有回流冷凝管的250mL的三口燒瓶中加入丙酸,再加入丙酸質量1.5%的二甲基甲醯胺,在回流冷凝管的一段插入尾氣吸收裝置,攪拌、升溫至70-75℃,再緩慢滴加氯化亞碸,丙酸與氯化亞碸的摩爾比為1:1.5。待反應完成後,減壓除去過量的氯化亞碸及副產物,即得丙醯氯,對所製備的丙醯氯進行檢測,得無色的丙醯氯的純度大於98%,收率為97.0%,沸點為79℃,密度為1.06g/mL。
在裝有回流冷凝管的250mL的三口燒瓶中加入1.00g N,N-二(2-氯乙基)-1,4-苯二胺、20.0mL乾燥的二氯甲烷和1.2mL的三乙胺,冰水浴冷卻,攪拌下滴加摩爾比為1:4的丙醯氯和二氯甲烷的混合溶液15.0mL,滴加時間約為1小時,其中,N,N-二(2-氯乙基)-1,4-苯二胺與丙醯氯的物料配比為1:2,滴加結束後撤掉冰水浴,常溫反應8小時,反應結束後使用30mL的水洗滌反應液,無水硫酸鈉乾燥,常壓蒸餾,濾餅用柱層析(V(石油醚:乙酸乙酯)1∶2)純化得暗紅色晶體,即為最終產物,產物收率為91.1%。
實施例3
1.1 N,N-二(2-羥乙基)-4-硝基苯胺的合成
向250mL三口燒瓶內分別加入摩爾比為1:1.4:0.7的4-氯硝基苯、二乙醇胺和碳酸鉀,最後加入質量濃度為1%的CuSO4溶液,且質量濃度為1%的CuSO4溶液與4-氯硝基苯的用量比為0.2mL:2g,混合均勻;加熱升高溫度,當溫度達到82℃時,向反應液中加入甲苯,且甲苯與4-氯硝基苯的用量比為1mL:3g;繼續升高溫度控制反應溫度為120℃,使用薄層色譜法跟蹤反應進程,恆溫攪拌反應7.5h。待反應完全後,蒸餾去除甲苯,將蒸餾後的母液倒入50℃的熱水中,劇烈攪拌,靜置過夜後抽濾除去水層得粗品,粗品用無水乙醇重結晶,得黃色針狀晶體N,N-二(2-羥乙基)-4-硝基苯胺。
1.2 N,N-二(2-氯乙基)-4-硝基苯胺的合成
稱取2.70g(0.012mol)黃色針狀晶體N,N-二(2-羥基乙基)-4-硝基苯胺,溶於60.0mL乾燥的二氯甲烷中,然後向反應瓶中加入1.1mL的三乙胺;取0.024mol二氯亞碸溶於20.0mL二氯甲烷溶液,室溫攪拌下將該溶液滴加到反應瓶,滴加的時間為l.5h,滴加完成後,加熱至45℃,回流,並在回流的狀態下攪拌反應2h。反應完全後靜置冷卻,用30.0mL乾燥的二氯甲烷稀釋,並分別用20.0mL的飽和碳酸氫鈉溶液洗滌三次,合併有機層用無水硫酸鈉乾燥,減壓蒸餾除去溶劑,得N,N-二(2-氯乙基)-4-硝基苯胺的粗品。粗品用無水乙醇重結晶得黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺2.99g,收率95.0%。
1.3 N,N-二(2-氯乙基)-1,4-苯二胺的合成
稱取4mmol黃色針狀晶體N,N-二(2-氯乙基)-4-硝基苯胺溶於200.0mL濃鹽酸中,然後加入12mmol氯化亞錫,氮氣保護下回流待溶液顏色變為無色透明時停止反應。用0℃的冰水浴冷卻,加入20.0mL的水稀釋,用濃氨水調pH至7.5~8.0,用乙酸乙酯萃取3次,無水硫酸鈉乾燥,減壓蒸除溶劑,即得。由於新生成的化合物具有化學活性比較高的基團氨基,其化學結構不穩定易被氧化,因此在用乙酸乙酯萃取後,向其溶液中加入過量的濃鹽酸,製備其鹽酸鹽低溫避光備用,待應用時加鹼萃取即可使用。
1.4式Ⅰ的芳胺氮芥衍生物N,N-二(2-氯乙基)-N′-丙醯基-1,4-苯二胺的合成
首先合成丙醯氯:在裝有回流冷凝管的250mL的三口燒瓶中加入丙酸,再加入丙酸質量1.5%的二甲基甲醯胺,在回流冷凝管的一段插入尾氣吸收裝置,攪拌、升溫至70-75℃,再緩慢滴加氯化亞碸,丙酸與氯化亞碸的摩爾比為1:1.5。待反應完成後,減壓除去過量的氯化亞碸及副產物,即得丙醯氯,對所製備的丙醯氯進行檢測,得無色的丙醯氯的純度大於98%,收率為97.0%,沸點為79℃,密度為1.06g/mL。
在裝有回流冷凝管的250mL的三口燒瓶中加入1.00g N,N-二(2-氯乙基)-1,4-苯二胺、20.0mL乾燥的二氯甲烷和1.2mL的三乙胺,冰水浴冷卻,攪拌下滴加摩爾比為1:6的丙醯氯和二氯甲烷的混合溶液15.0mL,滴加時間約為2小時,其中,N,N-二(2-氯乙基)-1,4-苯二胺與丙醯氯的物料配比為1:2.5,滴加結束後撤掉冰水浴,常溫反應14小時,反應結束後使用30mL的水洗滌反應液,無水硫酸鈉乾燥,常壓蒸餾,濾餅用柱層析(V(石油醚:乙酸乙酯)1∶3)純化得暗紅色晶體,即為最終產物,產物收率為91.1%。
對上述實施例1製備的N,N-二(2-羥乙基)-4-硝基苯胺進行元素分析,結果如表1所示。
表1 N,N-二(2-羥乙基)-4-硝基苯胺的元素分析結果
對N,N-二(2-羥乙基)-4-硝基苯胺採用溴化鉀壓片法進行紅外分析,結果如圖1所示,產物在3282cm-1處出現強吸收峰,屬於-OH的伸縮振動所產生的吸收峰;2906cm-1處出現吸收峰,歸屬亞甲基及中C-H的伸縮振動所產生的吸收峰;1600cm-1、1521cm-1、1483cm-1吸收峰是苯環的骨架振動所產生的吸收峰;1203cm-1處出現的吸收峰屬C-N振動吸收峰;819cm-1出現的吸收峰表明為苯環的對二取代C-H彎曲振動引起的吸收峰;1326cm-1處出現的吸收峰為硝基的伸縮振動所產生的吸收峰。綜合以上元素分析及紅外圖譜分析結果確定所合成的化合物為目標化合物N,N-二(2-羥乙基)-4-硝基苯胺。
對上述實施例1製備的N,N-二(2-氯乙基)-4-硝基苯胺進行元素分析,結果如表2所示。
表2元素分析結果
對N,N-二(2-氯乙基)-4-硝基苯胺採用溴化鉀壓片法進行紅外分析,結果如圖2所示,對比圖1和圖2可知,產物在3282cm-1處未出現-OH的伸縮振動吸收峰,且圖2較圖1在752cm-1及694cm-1處出現了比較強的吸收峰,歸屬為C-Cl伸縮振動產生的吸收峰;同樣在2962cm-1處出現了亞甲基中C-H的伸縮振動吸收峰;1591cm-1、1510cm-1、1494cm-1處的吸收峰是苯環的骨架振動產生的吸收峰;827cm-1處出現的吸收峰表明苯環的對二取代C-H健彎曲振動的吸收峰;1317cm-1處出現的吸收峰為硝基的N-O健伸縮振動產生的吸收峰。綜合以上元素分析及紅外圖譜分析結果確定所合成的化合物為目標化合物N,N-二(2-氯乙基)-4-硝基苯胺。
對上述實施例1製備的N,N-二(2-氯乙基)-1,4-苯二胺進行元素分析,結果如表3所示。
表3元素分析結果
對N,N-二(2-氯乙基)-1,4-苯二胺採用溴化鉀壓片法進行紅外分析,結果如圖3所示,對比圖3和圖2可知,圖3在3562cm-1及3442cm-1處出現了兩個比較弱的吸收峰,該峰歸屬伯胺N-H的伸縮振動吸收峰;2962cm-1處強吸收峰,歸屬亞甲基的C-H伸縮振動吸收峰;1571cm-1、1496cm-1、1458cm-1處的吸收峰歸屬苯環的骨架振動所產生的吸收峰;819cm-1處的吸收峰表明為苯環的對二取代C-H健彎曲振動的吸收峰;1431cm-1處出現的吸收峰歸屬C-N伸縮振動吸收峰;圖中在2586cm-1及2333cm-1處出現的兩個中強吸收峰,歸屬為胺鹽N-H振動吸收峰。綜合元素分析結果及紅外圖譜分析結果確定所合成的化合物為目標化合物N,N-二(2-氯乙基)-1,4苯二胺。
對上述實施例1製備的式Ⅰ的芳胺氮芥衍生物進行元素分析,結果如表4所示。
對上述式Ⅰ的芳氮芥衍生物採用溴化鉀壓片法進行紅外分析,如圖4所示,其主要譜帶的歸屬結果如表5所示。
表4式Ⅰ的芳胺氮芥衍生物的元素分析結果
表5式Ⅰ的芳氮芥衍生物的紅外主要譜帶分析結果
對上述式Ⅰ的芳氮芥衍生物進行核磁分析,用TMS作內標,DMSO作為溶劑,其核磁圖譜見圖5,其核磁氫譜數據和結構指派結果見表6。
由圖4和表5可知,在3277cm-1處出現的吸收峰屬於醯胺基的-NH伸縮振動所產生的吸收峰;1651處出現強的吸收峰歸屬C=O的伸縮振動所產生的吸收峰;2982cm-1處出現的吸收峰為甲基亞甲基C-H伸縮振動的吸收峰;1600~1515處出現的三組吸收峰歸屬芳環的骨架振動所產生的吸收峰;777及663cm-1處出現吸收峰歸屬C-Cl伸縮振動吸收峰;由此可以初步判斷合成了目標化合物。再由圖5和表6的核磁譜圖可知在δH0.97~2.28之間出現了(-COCH2CH3)中甲基、亞甲基的兩組特徵吸收峰;δH3.05~3.69之間同樣出現了N,N-二(2-氯乙基)胺中CH2的特徵峰;δH6.67~7.42之間出現了兩組苯環上氫的特徵吸收峰;δH9.62處的特徵峰為(NHCO)中氫的特徵吸收峰。表5的元素分析結果理論值與實測值無顯著性差異。因此由紅外譜圖、核磁譜圖及元素分析結果可以確定所合成的化合物為式Ⅰ的芳氮芥衍生物N,N-二(2-氯乙基)-N』-丙醯基-1,4-苯二胺。
表6式Ⅰ的芳氮芥衍生物的核磁氫譜數據分析結果
本發明提出的N,N-二(2-氯乙基)-1,4-苯二胺在低氧環境下有較好的細胞毒性,氨基的供電子作用使氮芥的氮原子上電子云密度增大,提高了氮芥的烷化作用。而腫瘤細胞中存在許多低氧細胞,正常細胞中的細胞相對是富氧的,如果藥物只在低氧環境中有細胞毒性,那麼這種藥物就對腫瘤細胞具有選擇性。本發明以N,N-二(2-氯乙基)-1,4-苯二胺為藥效基團,以4-氯硝基苯,二乙醇胺為起始原料在鹼性物質的存在下,通過親核取代反應一步合成了N,N-二(2-羥乙基)-4-硝基苯胺,所得化合物與二氯亞碸在三乙胺催化作用下,合成了芳胺氮芥類的主體化合物N,N-二(2-氯乙基)-4-硝基苯胺;以二價錫作為還原劑使N,N-二(2-氯乙基)-4-硝基苯胺4位的硝基還原為氨基得到氮芥類藥物的藥效體N,N-二(2-氯乙基)-1,4-苯二胺;以丙醯氯對N,N-二(2-氯乙基)-1,4-苯二胺進行結構修飾,使得本發明的芳胺氮芥衍生物具有增強氮芥治療指數的前提下,又可以有效降低氮芥的毒副作用,同時兼具殺菌、消炎的療效,以達到降低病人化療後因免疫力下降而引起的併發症的風險。
雖然,本說明書中已經用一般性說明及具體實施方案對本發明作了詳盡的描述,但在本發明基礎上,可以對之作一些修改或改進,這對本領域技術人員而言是顯而易見的。因此,在不偏離本發明精神的基礎上所做的這些修改或改進,均屬於本發明要求保護的範圍。