RNASTITCH測序:用於直接映射細胞中RNA:RNA相互作用的測定的製作方法
2023-07-18 20:51:16 2
相關申請的交叉引用
本申請要求2014年9月22日提交的美國臨時專利申請62/053,615的優先權的權益。上述申請的全部公開內容通過引用全部明確地併入本文。
關於聯邦贊助的r&d的聲明
本發明是在美國國立衛生研究院頒發的授權號nihdp2-od007417下由政府支持完成。政府對本發明具有特定的權利。
序列表、表格或電腦程式列表的參考
本申請與電子格式的序列表一起提交。所提供的序列表的名稱為ucsd089-001wo.txt,於2015年9月18日創建,文件大小為11kb。電子格式的序列表中的信息通過引用整體併入本文。
提供了用於鑑定細胞中彼此相互作用的rna的方法和組合物。
背景技術:
目前,沒有可以直接快速測定細胞類型中基本上所有rna-rna相互作用的有效方法。已有兩種方法來部分實現這一目標,但二者均有缺點。如hits-clip和clash等技術可以檢測許多mirna的靶點。然而,這兩種方法都集中在僅包含小部分rna的mirna。因此,這些技術不能揭示大多數rna-rna相互作用。此外,每項技術還有其他缺點。例如,不能直接從hits-clip推導出mirna與其靶mrna的直接配對。換句話說,hits-clip不直接通知哪個mirna調控哪些mrna(沒有一對一的信息)。
最近稱為clash(交聯、連接和混合測序)的方法可以允許直接觀察mirna-靶配對。然而,與測序讀段(read)的數量相比,相互作用的數量仍然很小:只有2%的測序讀段是嵌合的,98%仍然是單一讀段。這需要更深入的測序覆蓋或多個樣品的製備以獲得足夠的mirna-mrna相互作用的覆蓋。
技術實現要素:
本發明的一些實施方案在以下編號的段落中提供:
1、一種用於產生包含在細胞中彼此相互作用的rna的嵌合rna的方法,所述方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。
2、根據段落1所述的方法,其中所述rna與蛋白質的交聯在完整細胞上或在細胞裂解物中進行。
3、根據段落1或2任一項所述的方法,其中所述交聯包括uv交聯。
4、根據段落1-3任一項所述的方法,其還包括將所述蛋白質與有助於將所述蛋白質在表面上固定的試劑相關聯。
5、根據段落4所述的方法,其中促進固定的所述試劑包括生物素。
6、根據段落1-5任一項所述的方法,其還包括將與所述相同蛋白質分子交聯的所述rna片段化。
7、根據段落6所述的方法,其中所述片段化包括在有助於所述rna部分消化的條件下,將與所述相同蛋白質分子交聯的所述rna與rnase接觸。
8、根據段落1-7任一項所述的方法,其還包括將與所述相同蛋白質分子交聯的所述rna連接至有助於所述rna的回收的試劑。
9、根據段落8所述的方法,其中所述連接包括將所述rna的末端連接至所述試劑。
10、根據段落9所述的方法,其中有助於所述rna的回收的所述試劑包括核酸。
11、根據段落10所述的方法,其中所述核酸包括其上具有生物素的核酸。
12、根據段落11所述的方法,其中所述其上具有生物素的核酸與所述rna的所述末端的所述連接包括:在將與相同蛋白質分子交聯的所述rna連接在一起以形成嵌合rna之前,將其上具有生物素的所述核酸連接至所述rna的5'末端。
13、根據段落12所述的方法,其還包括從所述嵌合rna的5'區域除去所述生物素。
14、根據段落1-13任一項所述的方法,其還包括回收所述嵌合rna。
15、根據段落1-14任一項所述的方法,其還包括片段化所述嵌合rna。
16、根據段落1-15任一項所述的方法,其中所述嵌合rna的所述片段化包括:在有助於所述rna部分消化的條件下,使所述嵌合rna與rnase接觸。
17、根據段落1-16任一項所述的方法,其還包括反轉錄所述嵌合rna以產生嵌合cdna。
18、根據段落1-17任一項所述的方法,其還包括測定所述嵌合rna或嵌合cdna中源自所述嵌合rna或嵌合cdna中每個rna的至少一部分序列。
19、根據段落1-17任一項所述的方法,其還包括鑑定存在於所述嵌合rna中的rna,從而鑑定在細胞中彼此相互作用的rna。
20、根據段落19所述的方法,其中鑑定出所述細胞中至少100個、至少500個、至少1000個或多於1000個rna-rna相互作用。
21、根據段落19所述的方法,其中鑑定出所述細胞中基本上所有的彼此相互作用的rna。
22、根據段落21所述的方法,其中鑑定出所述細胞中至少70%、至少80%、至少90%或超過90%的直接rna-rna相互作用。
23、根據段落19-22任一項所述的方法,其中在細胞中彼此相互作用的rna的鑑定包括使用自動測序裝置對所述嵌合rna進行序列讀段。
24、根據段落23所述的方法,其中在細胞中彼此相互作用的rna的鑑定包括從所有序列讀段中鑑定嵌合序列。
25、根據段落19-24任一項所述的方法,其還包括使用計算機將所述嵌合rna轉化為注釋的rna簇。
26、根據段落25所述的方法,其還包括使用由計算機執行的統計檢驗來鑑定所述rna簇之間的直接相互作用。
27、一種分離的複合體,其包括與蛋白質交聯的嵌合rna,其中所述嵌合rna包括在細胞中彼此相互作用的rna。
28、一種鑑定候選治療劑的方法,其包括:
使用根據段落1-26任一項所述的方法鑑定細胞中彼此相互作用的rna;和
評估試劑減少或增加所述rna的相互作用的能力,其中如果所述試劑能夠減少或增加所述rna的相互作用,則所述試劑是候選治療劑。
29、根據段落28所述的方法,其中所述試劑包括核酸。
30、根據段落28所述的方法,其中所述試劑包括化學化合物。
31、一種製備藥物的方法,其包括將使用根據段落28-30任一項所述的方法鑑定的試劑配製在藥學上可接受的載體中。
32、一種藥物,其使用根據段落31所述的方法製備。
33、一種用於產生包含在細胞中彼此相互作用的rna的嵌合rna的方法,所述方法包括將rna與蛋白質中間體和/或蛋白質複合體交聯並將與蛋白質中間體和/或蛋白質複合體交聯的rna連接在一起以形成嵌合rna,並且其中所述蛋白質複合體包括兩種以上的相互作用蛋白。
34、根據段落33所述的方法,其中rna與蛋白質中間體和/或蛋白質複合體的所述交聯在完整細胞上或在細胞裂解物中進行。
35、根據段落33或34所述的方法,其中所述交聯包括uv交聯。
36、根據段落33-35任一項所述的方法,其還包括將所述蛋白質中間體和/或蛋白質複合體與有助於所述蛋白質中間體和/或蛋白質複合體在表面上固定的試劑相關聯。
37、根據段落36所述的方法,其中有助於固定的所述試劑包括生物素。
38、根據段落33-37任一項所述的方法,其還包括將與至少一種蛋白質分子交聯的所述rna片段化。
39、根據段落38所述的方法,其中所述片段化包括在有助於所述rna部分消化的條件下將與蛋白質中間體和/或蛋白質複合體交聯的所述rna與rnase接觸。
40、根據段落33-39任一項所述的方法,其還包括將與蛋白質中間體和/或蛋白質複合體交聯的所述rna連接至有助於所述rna的回收的試劑。
41、根據段落40所述的方法,其中所述連接包括將所述rna的末端連接至所述試劑。
42、根據段落41所述的方法,其中有助於所述rna回收的所述試劑包括核酸。
43、根據段落42所述的方法,其中所述核酸包括其上具有生物素的核酸。
44、根據段落43所述的方法,其中其上具有生物素的所述核酸與所述rna的所述末端的連接包括在將與蛋白質中間體和/或蛋白質複合體交聯的所述rna連接在一起以形成嵌合rna之前將其上具有生物素的所述核酸連接至所述rna的5'末端。
45、根據段落44所述的方法,其還包括從所述嵌合rna的5'區域除去所述生物素。
46、根據段落33-45任一項所述的方法,其還包括回收所述嵌合rna。
47、根據段落33-46任一項所述的方法,其還包括片段化所述嵌合rna。
48、根據段落33-47任一項所述的方法,其中所述嵌合rna的所述片段化包括在有助於所述rna部分消化的條件下使所述嵌合rna與rnase接觸。
49、根據段落33-48任一項所述的方法,其還包括反轉錄所述嵌合rna以產生嵌合cdna。
50、根據段落33-49任一項所述的方法,其還包括測定所述嵌合rna或嵌合cdna中源自所述嵌合rna或嵌合cdna中每個rna的至少一部分序列。
51、根據段落33-49任一項所述的方法,其還包括鑑定存在於所述嵌合rna中的rna,從而鑑定在細胞中彼此相互作用的rna。
52、根據段落51所述的方法,其中鑑定出細胞中至少100個、至少500個、至少1000個或多於1000個rna-rna相互作用。
53、根據段落51所述的方法,其中鑑定出所述細胞中基本上所有的彼此相互作用的rna。
54、根據段落53所述的方法,其中鑑定出所述細胞中至少70%、至少80%、至少90%或超過90%的直接rna-rna相互作用。
55、根據段落51-54任一項所述的方法,其中在細胞中彼此相互作用的rna的鑑定包括使用自動測序裝置對所述嵌合rna進行序列讀段。
56、根據段落55所述的方法,其中在細胞中彼此相互作用的rna的鑑定包括從所有序列讀段中鑑定嵌合序列。
57、根據段落51-56任一項所述的方法,其還包括使用計算機將所述嵌合rna轉化為注釋的rna簇。
58、根據段落57所述的方法,其還包括使用由計算機執行的統計檢驗來鑑定所述rna簇之間的直接相互作用。
59、根據段落33-58任一項所述的方法,其中在細胞中彼此相互作用的所述rna與所述蛋白質中間體或蛋白質複合體中的不同蛋白質進行交聯。
60、一種分離的複合體,其包括與蛋白質中間體和/或蛋白質複合體交聯的嵌合rna,其中所述嵌合rna包括在細胞中彼此相互作用的rna,其中所述蛋白質複合體包括兩種以上的相互作用蛋白。
61、根據段落59所述的分離的複合體,其中所述嵌合rna包括與所述蛋白質中間體或蛋白質複合體中的不同蛋白質交聯的rna。
附圖說明
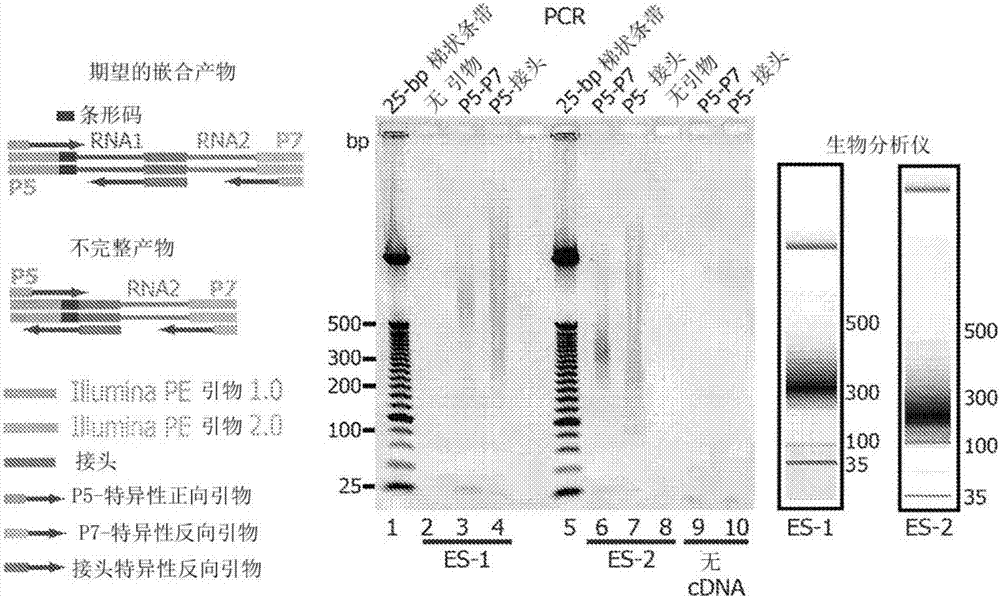
圖1.rnahi-c。(a)主要實驗步驟:1.將rna與蛋白質交聯,2.rna片段化和蛋白質生物素化(球代表生物素),3.固定化,4.連接生物素化的rna接頭(鏈上的球是接頭上的生物素),5.極度稀釋條件下的鄰位連接,6.rna純化和反轉錄,7.生物素下拉,8.構建測序文庫。嵌合rna示意圖中所示的是具有p5特異性引物、pr特異性引物和rna1之間的條形碼、rna1和rna2之間的接頭特異性反向引物以及p7區域的期望嵌合產物。在所示的不完整的產物中,p5區域與條形碼相鄰,條形碼位於p5區域和接頭之間,然後是rna2區域和p7區域。(b)rna1-接頭-rna2嵌合體的pcr驗證,其預期從p5測序引物到接頭為91bp以上以及p5至p7測序引物為200bp以上。不包括rna1的將產生從p5到接頭的91bp產物。不包括rna2的將產生從p5到接頭以及從p5到p7的類似大小的產物。每個泳道頂部標記有pcr引物。測序文庫的尺寸分布也由生物分析儀(bioanalyzer)評估。如從左至右的期望嵌合產物所示的是p5特異性正向引物、條形碼、rna1、接頭(與接頭特異性引物互補)、rna2和p7。如不完全產物所示,是p5,條形碼,接頭,rna2和p7。(c)映射到基因組的rnahi-c數據。trim25和snora1rna的連接在es-1和es-2文庫中由46個雙端讀段實驗支持。agoclip-seq:小鼠es細胞的agohits-clip(geo:gsm622570)。小rna-seq:具有用酶切割產生的3'羥基的小rna測序(geo:gsm945907)。(d)rna相互作用物組的大型模塊。沒有顯示涉及4個以下相互作用的rna的小模塊。沒有顯示涉及snorna、snrna和trna的相互作用。列表中的大部分序列是mrna,其餘的是假基因(fpl30=ps3,gm16580,gm12715,gm13226,rp128-ps3,fpl28-ps1,rps16-ps2,gm4707,gm13340,gm13408,gm15590,grl2,gm11400,gm17087,gm15725,gm12346,gm11478),lincrna(gm16869,malat1,snhg7,gm16702,4930417h01rik),mirna(mir5100,mir692-1,mir692-2b,ac117657,mir5099)和反義rna(gm15444)。
圖2.rna相互作用位點。(a)代表不同相互作用(虛線)的多重rnahi-c讀段,重疊在eef1a1基因的特定區域上。(b)通過重疊讀段的「峰」來查找相互作用位點。峰1和2是rna2,峰3和4是rna2。(c)不同類型的rna基因和轉座子中相互作用位點的分布。(d)兩個rna(淺灰色,左側)的相互作用位點之間和隨機改組鹼基(白色,右側)之間的結合能(δg,kcal/mol)的分布。來自wilcoxon秩檢驗的p值標記在每個圖的底部。(e)通過平均phylop得分測量的保守水平,在連接的rna片段的連接接合點(黑條,x軸上的0位置)達到峰值。對照:隨機選擇的基因組區域的保守水平。如圖所示,圖中左側的數據表示rna1,右側數據表示rna2。
圖3.rna結構。(a)解析rna鄰近位點的示意圖。核酸示意圖上的指示箭頭:rnasei切割位點。(b)映射到snora73的「切割和連接」產物。垂直色條:支持一對鄰近位點的一組讀段對。鄰近位點上的數字對應於圖3中e和f圖中的序列上的數字。(c)rnasei切割的密度。鄰近位點上的數字對應於圖3中e和f圖中的序列上的數字。(d)rna的任何兩個位置之間的連接頻率的熱圖。每個彩色圓圈對應於圖a中的垂直色條,並且表示一對鄰近位點。(e)承認的二級結構中單鏈區域和推斷的鄰位位點的足跡。(f)由於蛋白質輔助的rna摺疊,一對推斷的鄰位位點,其不受基於序列的二級結構的支持,在體內物理上接近。
圖4.所示為基於逐步測序以映射rna-rna相互作用的技術。
圖5.計算部分的工作流程。(a)用於鑑定嵌合rna序列的流程圖。如主要序列的插入框所示,是「無接頭」、「僅接頭」、「僅反向」、「僅正向」和「配對」的序列。如圖所示,無接頭的序列具有:1)5'索引,2)5'索引、第1部分和第2部分,3)5'索引、第1部分,和3)5'索引和第2部分。如圖所示,僅接頭序列具有5'索引和第2部分。如圖所示,「僅反向」具有5'索引、接頭和第2部分。如圖所示,「僅正向」具有5'索引和接頭。如圖所示,「配對」具有5'索引、第1部分、接頭和第2部分。(b)如何鑑定大量嵌合rna支持的rna-rna相互作用的圖示。如頂圖示出r1中的區段,下圖示出r2中的區段。如圖所示,它們在嵌合rna中配對。
圖6.初步結果。(a)嵌合cdna文庫的尺寸分布。注意128bp是引物序列。(b)不同類型rna之間相互作用的比例。(c)將18個連接的rna對映射到snora1和trim25。映射的位點與agoclip-seq數據(gsm622570)相符。(d)在導向分化過程中snora1和trim25的反向相關性。如圖所示,trim25在第4天從約35個rna-seqrpkm減少到約5個,而snora1從第0天到第6天增加。
圖7.用於構建測序文庫的環化策略。該圖詳細說明了rnahi-c程序的步驟8。(圖7a)將反轉錄(rt)適配子(adaptor)連接至rna的3'末端。該rt適配子與一部分rt引物互補,rt引物還含有用於p5測序引物的適配子,10nt條形碼和bamhi限制性位點。環化後,將含有bamhi位點的dna寡核苷酸與rt引物區雜交,提供用於bamhi消化的雙鏈底物。通過截短的pcr引物dp5和dp3擴增線性化的ss-cdna,得到~100ng的ds-cdna,然後將其變性並再退火。使用雙鏈特異性核酸酶(dsn)來耗盡源自rrna的cdna。dsn選擇性地除去在再退火過程中較早形成的ds-cdna。源自rrna的cdna應該是豐度較高的,因此比其他cdna更快再退火。通過illuminapcr引物pe1.0和2.0再次pcr擴增經dsn處理的產物以產生適於測序的文庫。將基於dsn的rrna去除應用於es-1。對es-2進行基於抗體的rrna去除策略,未在該圖中描繪。最終顯示的是p5、條形碼、rna1、適配子、rna2和p7的產物(圖7b)。
圖8.rnahi-c樣品的描述。「讀段對的總數#」是每個樣品的雙端順序讀段的數量。「rna1-接頭-rna2」形式的非重複讀段對#」是生物信息學流程的步驟4解析嵌合cdna的輸出中的雙端讀段的數量。
圖9.優化用於第一次片段化的rnasei濃度。通過加入等體積的2x蛋白酶k緩衝液(100mmtris-hclph7.5,100mmnacl,2%sds,20mmedta)和1:5體積的20mg/ml的蛋白酶k(neb),並在55℃下孵育2小時,然後進行苯酚:氯仿處理和乙醇沉澱,來從rnasei處理的es細胞裂解物中純化rna。每毫升細胞裂解物的rnasei的量為:0u(樣品1,圖9a),2.5u(樣品2(圖9b)),3.3u(樣品3,圖9c),5u(樣品4,圖9d)和12.5(樣品5,圖9e)。選擇產生500-1000ntrna片段(樣品4)的5.0urnasei/ml裂解物的濃度用於rnahi-c步驟2。
圖10.測試珠上接頭連接的效率。固定的rna用rnasei消化,然後與生物素標記的rna接頭連接(1)。連接和蛋白酶k消化除去蛋白質後,純化和定量rna(1.3μg)(2)。然後將純化的rna進行鏈黴親和素-生物素下拉以選擇連接至生物素標記的接頭的rna(3)。洗滌和洗脫結合至鏈黴親和素珠的rna並乙醇沉澱後,收集到0.22μgrna。同時,生物素標記的rna接頭進行相同的鏈黴親和素-生物素下拉,洗脫和乙醇沉澱(4)。假設步驟3和4中生物素下拉、rna洗脫和乙醇沉澱的效率相同,約19.6%(1.96μg/10.0μg),估計連接效率(0.22μg/19.6%)/1.3μg=86%。
圖11.rnahi-c程序的不同步驟的rna尺寸分布。只有es間接和mef樣品才有足夠的中間產物用於該回顧性分析。在連結到鏈黴親和素珠之前的mef(泳道1)和es-間接(泳道2)的裂解物中,在固定化後的上清液(泳道3和4)中,以及在鄰位連接後固定在珠上(es-間接:泳道5,mef:泳道6)的rna的尺寸分布。rna在2xrna上樣染料(neb)中在70℃變性5分鐘,在1.5%天然瓊脂糖凝膠上電泳並用sybrgold(invitrogen)染色。
圖12.構建測序文庫的pcr循環數的優化。在rnahi-c程序的步驟8中,使用截短形式的illuminapcr測序引物(dp5,dp3),用12個循環的pcr來預擴增es-1樣品的單鏈cdna。pcr產物用1.8×spriselect珠純化,其在通過雙鏈特異性核酸酶耗盡由rrna合成的cdna之前產生86ng雙鏈dna。使用nebnexthigh-fidelity2xpcrmastermix(neb)和illuminapeprimer1.0和2.0,以不同的pcr循環數(12,15,18),擴增來自總計22μl的rrna耗盡的雙鏈cdna的1μl等分試樣。在6%tbepage凝膠上測定pcr產物並用sybrgold(invitrogen)染色。基於凝膠結果,然後用11個循環的pcr擴增18μl原始rrna耗盡的雙鏈dna,以產生測序文庫。
圖13.rnahi-c文庫的比較。(圖a-b)將接頭的5'末端(rna1)和3'末端(rna2)上的讀段片段分別作為兩個rna-seq實驗分析。所有已知rna在es-1和es-2樣品之間的讀段計數分布(fpkm)的以對數標度計的散點圖。r:皮爾遜相關。s:斯皮爾曼相關。(圖13c)每個樣品的fpkm的分層聚類。
圖14.rna-hic工具的在線文檔。該在線資源(http://systemsbio.ucsd.edu/rna-hi-c)包括分析和可視化工具、使用示例、示例輸出文件和圖形的詳細描述。還提供了一些工具作為應用程式編程接口(api)。
圖15.用於分析rnahi-c數據的計算流程。(a)從雙端測序讀段中移除pcr重複(步驟1)。基於4nt實驗條形碼('xxxx',步驟2)分離多重樣品。'n':隨機條形碼的核苷酸。'x':實驗條形碼的核苷酸。(b)如果可能,每對正向(read1)和反向(read2)讀段被用於在輸入測序文庫中回收cdna。(c)基於rna片段和接頭序列的配置對回收的cdna進行分類(步驟4)。提供rna1-接頭-rna2型cdnas作為輸出。(d)將rna1部分和rna2部分分別映射到基因組。輸出是rna1和rna2唯一地映射到基因組的cdna。(e)基於關聯測試鑑定rna-rna相互作用。如圖所示,簇1和簇2具有rna1,簇3和簇4具有rna2。
圖16.rna-hic-工具的可視化能力。(a-b)rna內rna(a)和rna間rna(b)相互作用中rna相互作用位點的詳細視圖。將含有兩個相互作用的rna的兩個基因組區域平行繪製(圖b)。將每個rna1-接頭-rna2型嵌合rna作圖,其中將rna1和rna2片段映射到相應的基因組區域,通過表示接頭的斜線連接。塊表示重疊的rnahi-c讀段的「峰」,它們是候選rna相互作用位點。連接兩個rna相互作用位點的半透明多邊形代表了強相互作用。(c)rna-rna相互作用的全局視圖。rna1和rna2片段的讀段密度分別顯示在染色質細胞帶表意文字內的陰影區域。每個鑑定的rna-rna相互作用顯示為連接兩個rna的基因座位點的曲線,並通過相互作用的rna的類型著色。
圖17.具有mirna樣相互作用的snorna。(a)rnahi-c與小rna-seq(gsm945907)和agohits-clip(gsm622570)的比較。在小rna-seq和agohits-clip中參與rnahi-c鑑定的相互作用的每種類型的rna的平均fpkm以對數標度顯示。rnahi-c鑑定的相互作用中的mirna和snorna在小rna-seq和agohits-clip中均富集。如圖17的a圖所示,該圖表示為代表小rna-seq數據的柱超過代表h1ts-clip數據的柱。(b)每對相互作用的snorna和mrna之間基因表達的相關性分布。由ago結合的相互作用的snorna-mrna對(深灰色)(由agohits-clip定義)與不被ago結合的對(淺灰色)相比更負相關(p值=4.18-5,kolmogorov-smirnov檢驗)。如圖所示,ago結合峰出現在約0.075、0.25、0、-0.5和-1相關。(c)通過雜交能測量的相互作用的rna的鹼基配對。由ago結合的snorna-mrna對(與agohits-clip相交,左側)表現出比不被ago結合的(右側)更強的雜交能量(p值<2.2-16,wilcoxon符號秩檢驗)。所有這些相互作用顯示出比具有隨機改組序列的那些更強的雜交能。如圖所示,深灰色表示「真實」,淺灰色表示「隨機」。(d)在小rna-seq和agohits-clip中與mrna的utr區相互作用的snorna富集。snorna和mrna編碼區(左)之間的相互作用的總數(y軸)分解為在小rna-seq和agohits-clip中檢測到的,僅在小rna-seq中檢測到的,僅在hits-clip中檢測到的,以及在兩個數據集中都未檢測到的。snorna和mrnautr之間的相互作用相似地分解(右)。如左側柱形圖所示,頂部是小rna和clip,接著是clip數據、小rna和「兩者都不」。
圖18.rnahi-c和小rna-seq和agohits-clip之間的比較。與小rna-seq、agohits-clip和兩者交叉的rnahi-c鑑定的相互作用的百分比。rnahi-c相互作用按照參與rna的類型進行分類,並按與hits-clip的重疊對分類進行排名。misc_rna:混雜的rna(miscellaneousrna),包括rnase_mrp,7skrna等。新:未注釋的rna。如圖所示,數據從上到下分為「與兩者重疊」,「與小rna-seq」重疊,以及「與hits-clip重疊」的數據。
圖19.酶處理的snora14和mcl1mrna之間的相互作用。(a)與小rna-seq交叉的snora14上的rnahi-c鑑定的相互作用位點,表明snora14rna被酶處理成較短的形式(峰上的突出顯示區域,第2行)。這種酶促處理的小rna對應於snora14髮夾的末端(二級結構上的突出顯示區域),以及mcl1的3'utr的反義(snoara14序列上方(b)中的突出顯示區域))。(c)在es細胞分化為內中胚層細胞期間從snora14rna和mcl1mrna處理的小rna的表達水平。如圖所示,mcl1從第0天到第6天下降,而snora14從第0天到第6天增加。
圖20.讀段計數和fdr的分布以及與基因表達的關係。(a)映射到每對rna的讀段對數量的分布。(b)fisher精確檢驗中每個rna對的fdr分布。(c)映射到每個rna的rnahi-c讀段數(y軸)和fpkm(x軸)的散點圖。(d)與每個rna的相互作用相關聯的最小fdr(以負對數計)和該rna的fpkm的散點圖。fpkm值通過用針對mm9的bowtie2-2.2.4映射來自小鼠encode數據集encsr000cwc(e14小鼠es細胞的雙端rna-seq)[1]的原始讀段,然後用cufflink2.2.1進行處理而獲得。在encsr000cwc數據和rna-hi-c小鼠es細胞數據中發現的具有獨特ensemblid的所有基因都包含在圖(c)和(d)中。
圖21.在不同類型rna中鑑定的46,780個rna-rna相互作用的分布。從分析中用實驗方法(實驗步驟6.2)和生物信息學方法(分析步驟6)移除rrna。
圖22.rna-rna相互作用網絡的程度分布。節點數(rna)與其作為無標度網絡的特徵的log標度(a)的程度(相互作用數)成反比。從網絡(b)中刪除snrna、snorna和trna後,該屬性沒有改變。
圖23.不同類型基因和轉座子中相互作用位點的分布。新:未注釋的基因組區域。
圖24.rnahi-c鑑定的相互作用的rna之間的鹼基互補的實例。相互作用的rna的類型包括mrna-mrna(a),lincrna-mrna(b),假基因rna-mrna(c),mrna-ltr(d),line-mrna(e),mrna-mirna(f)。ltr和line代表轉座子轉錄物。將rna的3'末端連接至第二個rna的序列的左側的曲線表示接頭位置。支持每個相互作用的連接的嵌合rna的數量在曲線旁邊的括號中給出。δg:雜交能。改組:隨機改組鹼基的平均雜交能。
圖25.相互作用的rna的保守水平。相互作用由rna類型分類。對於每種類型的相互作用,保守水平與以rna連接接合點(x軸上為0位)為中心的基因組區域(1000bp)的平均phylop得分近似。將相同長度的隨機基因組區域的保守水平繪製為對照。圖的底部是rna1-接頭-rna2嵌合rna的rna1(右)和rna2(左)片段的圖示。虛線:接頭。如圖所示,圖25a為具有mrna的結構,圖25b為具有line的結構,圖25c為具有ltr的結構。
圖26.保守水平的比較。通過相互作用位點每個核苷酸的平均phylop得分(y軸)定量保守水平。為了調整外顯子、內含子和utr的保守性差異,將注釋的外顯子、內含子和utr(稱為基因組特徵)中的相互作用位點(配對柱左側的柱)與具有相同的基因組特徵的200,000個隨機取樣的基因組序列(配對柱右側的柱)進行比較。隨機取樣的基因組序列的大小與相互作用位點的大小具有相同的平均值和偏差。由單側雙樣品t檢驗計算p值。**:p值<10-12;*:p值<10-6。
圖27.rna酶i消化密度和單鏈區域的相關性(圖27a-d)。將通過在每個位置(y軸)處結束或開始的讀段片段數量測量的消化頻率與已知二級結構(frnadb資料庫v3.4)(x軸)進行比較。x軸上的括號表示雙鏈區域。在單鏈(ss)和雙鏈(ds)中每個位置結束或開始的讀段片段的總計數總結在右圖上。
圖28.分子內連接。(a)通過轉錄物的rnasei消化產生分子內(自身)連接,然後進行接頭連接和鄰位連接。因此,接頭兩側的兩個rna片段來自相同的rna分子。用嚴格的生物信息學標準鑑定了這些分子內連接事件,篩選出可能從連續轉錄物產生的雙端讀段。將只能通過切割和連接過程產生的雙端讀段用於rna結構分析。下圖:不同rna類型中分子內連接的分布。(b)各rna類型的分子內連接數(y軸)與轉錄物長度(x軸)。誤差條:平均值的標準偏差。顯示的是每個基因的長度超過1000nt的lincrna,每個基因小於10個自身連接的長度小於100nt的trna,每個基因超過100個自身連接的長度超過100nt的snorna,每個基因小於100個自身連接的長度超過100nt的snrna。(c)根據檢測到的分子內連接數(x軸)分類的lincrna和mrna基因的數量(陰影柱)和長度(盒型圖)。
圖29.snora14上的rnahi-c讀段。(a)映射到snora14的分子內連接產物。在黑色區域顯示的是連接接合點。陰影數字是接頭5'和3'處主要代表的連接接合點的位置。1-6、1-4和5-5位置的空間鄰近度與序列預測的二級結構(b)一致。箭頭指向在序列預測二級結構上彼此不接近的3-5個位置。
圖30.產生結構穩定轉錄物的推定的新基因。(a)rnahi-c預測的新基因的基因組位置和種間保守性。(b)分子內連接產物映射到該新基因。黑色區域:連接接合點。陰影數字:主要代表的連接接合點的位置。(c)從該推定基因產生的長(底)和短(頂)轉錄物的序列預測二級結構。每個鹼基上的rnasei消化的頻率(熱圖)與預測的單鏈區域(底部)相關。連接位置(箭頭)在測序的預測二級結構上接近。
圖31.mrna的一部分的推斷結構。將rnahi-c讀段對疊加在從gcn111基因的第27個外顯子的序列中預測的二級結構上。標記曲線分別對應於測序的嵌合rna的rna1和rna2部分。陰影曲線:接頭。陰影曲線上的黑色區域:連接接合點。指針表示rnasei切割位置。切割和連接過程交換兩個rna片段的5'-3'順序:在測序的嵌合cdna(插入)上交換mrna的5'片段(鹼基3122-33163,紅色)和3'片段(鹼基3164-3194,藍色)。這將不得不通過製圖適當地遮蔽。
圖32.用於在測序文庫中回收嵌合cdna的工作流程。使用局部比對來鑑定讀段對中的正向和反向讀段之間的任何重疊。使用四次本地比對(align1-aling4)來區分任何讀段對的四種類型可能的構型。輸出中包含三種類型(類型1-3)。類型1cdna短於100bp。類型2cdna位於100bp和200bp之間。類型3cdna長於200bp。作為質量控制,棄去短於100bp但沒有p5或p7測序引物已知序列的cdna(類型4)。每次比對表示為『local-align(seq1,seq2){m,m,o,e}』,其中『seq1』和『seq2』是兩個輸入序列『m』,『m』,『o』,『e』是匹配(match)、錯配(mismatch)、開放空位罰分(open-gappenality)和延伸空位罰分(extend-gappenality)的參數。每次比對的輸出(x)包括比對得分(scorex)、第一個(beginpos1_x,endpos1_x)和第二個序列(beginpos2_x,endpos2_x)中比對的開始位置和結束位置。
圖33.模擬分析。(a)cdna的預測長度(y軸)和真實長度的散點圖。預測長度大於200bp的cdna不包括在內,因為它們的準確長度無法預測。(b)預測rna對和模擬rna對之間的重疊。(c)每種類型的參與rna的預測rna對的靈敏度和特異性。
圖34.小鼠es細胞(a)和腦(b)的整個觀察到的rna-rna相互作用網絡的程度分布。節點數(rna)與其作為無標度網絡的特徵的log標度的程度(相互作用數)成反比。
定義
在下面的描述中,廣泛使用了許多術語。提供以下定義以便於理解本替代方案。
如本文所用,「一個/種(a)」或「一個/種(an)」可指一個/種或多於一個/種。
如本文所用,術語「約」指一數值,該數值包括用於確定數值的方法的固有的誤差變化,或實驗之間存在的變化。
本文所述的「核糖核酸」,「rna」是指與其在編碼、解碼、調控和表達基因中的作用有關的聚合物分子的核酸。在本文所述的一些實施方案中,rna可通過催化生物反應、控制基因表達或感知和傳遞對細胞信號的響應而在細胞內起積極作用。有幾種類型的rna。不受限制地,rna可以包括例如信使rna(mrna),lincrna,轉座子rna,假rna,調控rna,小核rna(snrna),小核仁rna(snorna),雙鏈rna,長非編碼rna(長ncrna或lncrna),微小rna(mirna),短幹擾rna(sirna),piwi相互作用rna(pirna)和其他類型的短rna。在一些實施方案中,提供了用於產生包含在細胞中彼此相互作用的rna的嵌合rna的方法。該方法可以包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,rna是信使rna(mrna),調控rna,小核rna(snrna),小核仁rna(snorna),雙鏈rna,長非編碼rna(長ncrna或lncrna),微小rna(mirna),短幹擾rna(sirna),piwi相互作用的rna(pirna)或本領域技術人員已知的其它類型的短rna。
如本文所述的「嵌合rna」是指rna複合體,其中rna複合體包含連接至相同蛋白質分子的連接的rna,並且rna彼此連接以形成該嵌合rna。在一些實施方案中,提供了用於產生包括在細胞中彼此相互作用的rna的嵌合rna的方法。該方法可以包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,rna是信使rna(mrna),調控rna,小核rna(snrna),雙鏈rna,長非編碼rna(長ncrna或lncrna),微小rna(mirna),短幹擾rna(sirna),piwi相互作用的rna(pirna),小核仁rna(snorna)或本領域技術人員已知的其它類型的短rna。在一些實施方案中,提供了分離的複合體,其中所述分離的複合體包括與蛋白質交聯的嵌合rna,其中所述嵌合rna包括在細胞中彼此相互作用的rna。
如本文所述的「交聯」或「交聯的」是指可將一種聚合物與另一種聚合物連接的鍵。交聯可通過共價鍵或離子鍵發生。在一些實施方案中,rna通過uv誘導的交聯與蛋白質交聯。使用紫外光照射蛋白質-核酸複合體(包含蛋白質和核酸的複合體、中間體蛋白質和核酸、或者蛋白質複合體和核酸)可以在核酸和與核酸緊密接觸的蛋白質之間形成共價鍵。在本文的一些實施方案中,rna通過uv輻射與蛋白質交聯。
交聯也可以通過使用接頭以及本領域技術人員已知的其它交聯方法來進行。在一些實施方案中,交聯可以通過使用探針將蛋白質連接在一起以及本領域技術人員已知的其它交聯方法來進行。交聯可用於聚合物合成化學以及生物科學。交聯可以通過各種條件引發的化學反應形成。不受限制地,可以例如通過加熱、壓力變化、ph變化、uv光、電子束曝光、γ輻射和/或本領域技術人員已知的其它類型的輻射來引發交聯。此外,也可以通過交聯試劑誘導交聯,產生導致兩種聚合物之間的交聯的化學反應。在本文所述的一些實施方案中,通過熱、壓力變化、ph變化、uv光、電子束曝光、γ輻射和/或本領域技術人員已知的其它類型的輻射來引發交聯。
交聯試劑可以包括但不限於胺-胺交聯劑,巰基(sulfhydryl)-巰基的交聯劑,胺-巰基交聯劑,巰基-碳水化合物交聯劑,光反應性交聯劑,化學選擇性連接交聯試劑,體內交聯試劑和羧基-胺交聯劑。在一些實施方案中,交聯試劑包括甲醛,dsg(二琥珀醯亞胺戊二酸酯),dss(二琥珀醯亞胺辛二酸酯),bs3(雙(磺基琥珀醯亞胺)辛二酸酯),tsat(三(琥珀醯亞胺)氨基三乙酸酯),bs(peg)5(peg化雙(磺基琥珀醯亞胺)辛二酸酯),bs(peg)9(peg化雙(磺基琥珀醯亞胺)辛二酸酯),dsp(二硫代雙(琥珀醯亞胺丙酸酯)),dtssp(3,3'-二硫代雙(磺基琥珀醯亞胺丙酸酯)),dst(二琥珀醯亞胺酒石酸酯),bsocoes(雙(2-(琥珀醯亞氨基氧基羰基氧基)乙基)碸),egs(乙二醇雙(琥珀醯亞胺琥珀酸酯)),磺基-egs(乙二醇雙(磺基琥珀醯亞胺琥珀酸酯)),dma(己二亞胺酸二甲酯),dmp(庚二亞氨酸二甲酯)dms(辛二亞氨酸二甲酯),dtbp(wangandrichard'sreagent),dfdnb(1,5-二氟-2,4-二硝基苯),bmoe(雙馬來醯亞胺乙烷),bmb(1,4-雙馬來醯亞胺丁烷),bmh(雙馬來醯亞胺己烷),tmea三(2-馬來醯亞氨基乙基)胺),bm(peg)2(1,8-雙馬來醯亞氨基二甘醇),bm(peg)3(1,11-雙馬來醯亞氨基-三甘醇),dtme(二硫代二馬來醯亞氨基乙烷),sia(琥珀醯亞胺碘乙酸酯),sbap(琥珀醯亞胺基3-(溴乙醯胺基)丙酸酯),siab(琥珀醯亞胺基(4-碘乙醯基)氨基苯甲酸酯),磺基-siab(磺基琥珀醯亞胺基(4-碘乙醯基)氨基苯甲酸酯),amas(n-α-馬來醯亞胺乙醯基-氧代琥珀醯亞胺酯),bmps(n-β-馬來醯亞胺基丙基氧代琥珀醯亞胺酯),gmbs(n-γ-馬來醯亞胺基丁醯基-氧代琥珀醯亞胺酯),磺基-gmbs(n-γ-馬來醯亞胺基丁醯基-氧代磺基琥珀醯亞胺酯),mbs(間馬來醯亞胺基苯甲醯-n-羥基琥珀醯亞胺酯),磺基-mbs(間馬來醯亞胺基苯甲醯-n-羥基磺基琥珀醯亞胺酯),smcc(琥珀醯亞胺基4-(n-馬來醯亞胺基甲基)環己烷-1-羧酸酯),磺基-smcc(磺基琥珀醯亞胺基4-(n-馬來醯亞胺基甲基)環己烷-1-羧酸酯),emcs(n-ε-馬來醯亞胺基己醯基-氧代琥珀醯亞胺酯),磺基emcs(n-ε-馬來亞胺基己醯基-氧代磺基琥珀醯亞胺酯,smpb(琥珀醯亞胺基4-(對馬來醯亞胺基苯基)丁酸酯)),磺基-smpb(磺基琥珀醯亞胺基4-(n-馬來醯亞胺基苯基)丁酸酯),smph(琥珀醯亞胺基6-((β-馬來醯亞胺基丙醯胺基)己酸酯)),lc-smcc(琥珀醯亞胺4-(n-馬來醯亞胺基甲基)環己烷-1-羧基-(6-醯氨基己酸酯)),磺基-kmus(n-κ-馬來醯亞胺基癸醯基-氧代磺基琥珀醯亞胺酯),spdp(琥珀醯亞胺基3-(2-吡啶基二硫代)丙酸酯),lc-spdp(琥珀醯亞胺基6-(3(2-吡啶基二硫代)丙醯胺基)己酸酯),磺基-lc-spdp(磺基琥珀醯亞胺基6-(3'-(2-吡啶基二硫代)丙醯胺基)己酸酯),smpt(4-琥珀醯亞胺氧基羰基-α-甲基-α(2-吡啶基二硫代)甲苯),peg4-spdp(peg化長鏈spdp交聯劑),peg12-spdp(peg化長鏈spdp交聯劑),sm(peg)2(peg化smcc交聯劑),sm(peg)4(peg化smcc交聯劑),sm(peg)6(peg化長鏈smcc交聯劑),sm(peg)8(peg化長鏈smcc交聯劑)(peg)12(peg化長鏈smcc交聯劑),sm(peg)24(peg化長鏈smcc交聯劑),琥珀醯亞胺基3-(2-吡啶基二硫代)丙酸酯(spdp),smcc,琥珀醯亞胺基反式-4-(馬來醯亞胺基甲基)環己烷-1-羧酸酯,bmph(n-β-馬來醯亞胺丙酸醯肼),emch(n-ε-馬來醯亞氨基己酸醯肼),mpbh(4-(4-n-馬來醯亞胺基苯基)丁酸醯肼),kmuh(n-κ-馬來醯亞胺十一酸醯肼),pdph(3-(2-吡啶基二硫代)丙醯肼),anb-nos(n-5-疊氮基-2-硝基苯甲醯基-氧代琥珀醯亞胺),磺基-sanpah(磺基琥珀醯亞胺基6-(4'-氨基-2'-硝基苯基氨基)己酸酯),sda(nhs-雙吖丙啶(diazirine))(琥珀醯亞胺基4,4'-疊氮戊酸酯),磺基-sda(磺基-nhs-雙吖丙啶)(磺基琥珀醯亞胺基4,4'-疊氮戊酸酯),lc-sda(nhs-lc-雙吖丙啶)(琥珀醯亞胺基6-(4,4'-疊氮戊醯胺基)己酸酯),磺基-lc-sda(磺基-nhs-lc-雙吖丙啶)(磺基琥珀醯亞胺基6-(4,4'-疊氮戊醯胺基)己酸酯),sdad(nhs-ss-雙吖丙啶)(琥珀醯亞胺基2-((4,4'-疊氮戊醯胺基)乙基)-1,3'-二硫代丙酸酯),磺基-sdad(磺基-nhs-ss-雙吖丙啶)(磺基琥珀醯亞胺基2-((4,4'-疊氮戊醯胺基)乙基)-1,3'-二硫代丙酸酯),atfb,se,4-疊氮基-2,3,5,6-四氟苯甲酸,琥珀醯亞胺酯,sda(nhs-雙吖丙啶)(琥珀醯亞胺基4,4'-疊氮戊酸酯),spb(琥珀醯亞胺基-[4-(補骨脂素-8-基氧基)]-丁酸酯),l-光-亮氨酸,l-光-甲硫氨酸,mannaz(四醯化n-疊氮基乙醯基甘露糖胺),galnaz(四醯化n-疊氮基乙醯基半乳糖胺),dcc(二環己基碳二亞胺),dylighttm550-膦,dylighttm650-膦,ez-linktm膦-peg3-生物素,ez-linktm膦-peg4-脫硫生物素,edc(1-乙基-3-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽),nhs(n-羥基琥珀醯亞胺),磺基-nhs(n-羥基磺基琥珀醯亞胺),磺基-nhs(n-羥基磺基琥珀醯亞胺),磺基-nhs(n-羥基磺基琥珀醯亞胺)或磺基-nhs(n-羥基磺基琥珀醯亞胺)。
如本文所述的「固定」是指捕獲分子,其中捕獲由特異於特定分子或標記的第一分子進行。在一些實施方案中,通過將捕獲分子附著在固體支持物上來進行固定。固體支持物可以是珠或柱。在一些實施方案中,固體支持物包含用於捕獲分子的鏈黴親和素分子,例如鏈黴親和素或其部分。在一些實施方案中,蛋白質在半胱氨酸殘基處被生物素化。
如本文所述的「片段化」可以指消化或打斷核酸。在本文所述方法的一些實施方案中,rna被酶片段化。rna降解可以通過許多類型的核酸酶進行。例如,核糖核酸酶(rnase)是一種可以催化rna降解成較小組分的核酸酶。rnase可以分為核糖核酸內切酶和核糖核酸外切酶。在一些實施方案中,提供了用於產生包括在細胞中彼此相互作用的rna的嵌合rna的方法,其中所述方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,rna與蛋白質的交聯在完整細胞上或在細胞裂解物中進行。在一些實施方案中,交聯包括uv交聯。在一些實施方案中,所述方法還包括將所述蛋白質與有助於所述蛋白質在表面上固定化的試劑相關聯。在一些實施方案中,有助於固定的所述試劑包括生物素。在一些實施方案中,蛋白質在半胱氨酸殘基處被生物素化。在一些實施方案中,該方法還包括將與相同蛋白質分子交聯的所述rna片段化。在一些實施方案中,所述片段化包括在有助於所述rna部分消化的條件下將與相同蛋白質分子交聯的所述rna與rnase接觸。
本文所述的「生物素」是指也稱為維生素h或輔酶r的水溶性維生素b。在本文所述的多個實施方案中,生物素可用於標記rna用於通過固體支持物如珠上的鏈黴親和素分子捕獲。在一些實施方案中,提供用於產生包括在細胞中彼此相互作用的rna的嵌合rna的方法,其中所述方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,rna與蛋白質的交聯在完整細胞上或在細胞裂解物中進行。在一些實施方案中,交聯包括uv交聯。在一些實施方案中,所述方法還包括將所述蛋白質與有助於所述蛋白質在表面上固定的試劑相關聯。在一些實施方案中,有助於固定的所述試劑包括生物素。在一些實施方案中,蛋白質在半胱氨酸殘基處被生物素化。在一些實施方案中,該方法還包括將與相同蛋白質分子交聯的所述rna片段化。在一些實施方案中,所述片段化包括在有助於所述rna部分消化的條件下將與相同蛋白質分子交聯的所述rna與rnase接觸。在一些實施方案中,所述方法還包括將與相同蛋白質分子交聯的所述rna連接至有助於所述rna的回收的試劑。在一些實施方案中,所述連接包括將所述rna的末端連接至所述試劑。在一些實施方案中,有助於所述rna回收的所述試劑包含核酸。在一些實施方案中,所述核酸包括其上具有生物素的核酸。在一些實施方案中,所述其上具有生物素的核酸與所述rna的所述末端的所述連接包括在將與相同蛋白質分子交聯的所述rna連接在一起以形成嵌合rna之前將其上具有生物素的所述核酸連接至所述rna的5'末端。在一些實施方案中,所述方法還包括從所述嵌合rna的5'區域除去所述生物素。在一些實施方案中,所述方法還包括回收所述嵌合rna。在一些實施方案中,所述方法還包括將所述嵌合rna片段化。
本文所述的「蛋白質」是指包含一個或多個多肽鏈的大分子。因此,蛋白質可以由肽組成,肽是由任何一個或多個胺基酸形成的肽(醯胺)鍵連接的胺基酸單體的鏈。蛋白質或肽可以含有至少兩個胺基酸,並且對能夠包含蛋白質或肽序列的胺基酸的最大數目沒有限制。不受限制地,胺基酸例如是精氨酸,組氨酸,賴氨酸,天冬氨酸,穀氨酸,絲氨酸,蘇氨酸,天冬醯胺,穀氨醯胺,半胱氨酸,胱氨酸,甘氨酸,脯氨酸,丙氨酸,纈氨酸,羥脯氨酸,異亮氨酸,亮氨酸,吡咯賴氨酸,甲硫氨酸,苯丙氨酸,酪氨酸,色氨酸,鳥氨酸,s-腺苷甲硫氨酸和硒代半胱氨酸。蛋白質還可以包含非肽組分,例如碳水化合物基團。碳水化合物和其他非肽取代基可以通過產生蛋白質的細胞加入到蛋白質中,並且將隨細胞類型而變化。不受限制地,蛋白質可通過催化代謝反應、dna複製、響應刺激和將分子從一個位置傳遞到另一個位置而在生物體內起作用。例如,蛋白質可以是酶,跨膜蛋白和抗體,用於轉運的小生物分子,受體或激素。在一些實施方案中,提供了用於產生包括在細胞中彼此相互作用的rna的嵌合rna的方法,其中所述方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,所述蛋白質是酶。在一些實施方案中,所述蛋白質參與轉運或催化代謝反應。
本文所述的「相互作用物組(interactome)」是指特定細胞中的分子相互作用的整個集合。該術語具體指分子之間的物理相互作用(例如蛋白質之間的,也稱作蛋白質-蛋白質相互作用),但也可以描述基因之間的間接相互作用(遺傳相互作用)的集合,如rna-rna相互作用或一個或多個rna和蛋白質分子之間的相互作用。在一些實例中,可以將相互作用物組以圖形展示出來。在一些實施方案中,本發明的方法和組合物在一次測定中基本上描繪了所有蛋白質輔助的rna-rna相互作用。在本文描述的一些實施方案中,已經應用了該方法來產生rna相互作用物組的第一幅全局圖。在一些實施方案中,從特定細胞產生相互作用物組。在一些實施方案中,細胞來自人。在一些實施方案中,細胞是癌細胞,腫瘤細胞,淋巴細胞或免疫細胞。在一些實施方案中,相互作用物組可用於確定或預測疾病途徑。
如本文所定義的「蛋白質複合體」是指一組或兩組以上的相結合的蛋白或多肽鏈,也可稱為「多蛋白複合體」。在一些實施方案中,提供了包括結合至蛋白質複合體的核酸的複合體。在一些實施方案中,所述核酸是rna。
本文定義的「蛋白質中間體」是指可以在過程或特定途徑期間彼此結合和解脫的蛋白質,並且還可以稱為「蛋白質結合中間體」。不受限制地,其中可見蛋白質中間體結合的實例可以包括轉錄、翻譯和代謝途徑等過程。不受限制地,蛋白質結合中間體的實例可包括聚合酶,核酸結合蛋白,rna識別動力蛋白(moticprotein),異質核糖核蛋白顆粒和本領域技術人員已知的其它蛋白質結合中間體。在一些實施方案中,提供了包含與蛋白質中間體結合的核酸的複合體。在一些實施方案中,所述核酸是rna。在一些實施方案中,蛋白質中間體與其它蛋白質中間體相互作用,從而形成蛋白質複合體,其中所述蛋白質複合體包含蛋白質中間體。
具體實施方式
本文公開了用於鑑定細胞中直接rna-rna相互作用的方法和組合物。在一些實施方案中,所述方法和組合物可用於鑑定細胞中至少約100個、至少約500個、至少約1000個或多於約1000個rna-rna相互作用。在一些實施方案中,所述方法和組合物可用於鑑定約100、約200、約300、約300、約500、約600、約700、約800、約900、約1000、約2000、約3000、約4000、約5000、約6000、約7000、約8000、約9000或約10,000個rna-rna相互作用或任意兩個上述這些值之間的任何其它數目的rna-rna相互作用。在其它實施方案中,所述方法和組合物可用於鑑定細胞中基本上所有的直接rna-rna相互作用。例如,方法和組合物可用於鑑定細胞中直接rna-rna相互作用的至少約70%、至少約80%、至少約90%或多於約90%。在一些實施方案中,所述方法和組合物可用於鑑定細胞中直接rna-rna相互作用的至少約70%、至少約80%、至少約90%或約100%,或者任意兩個上述值之間的任何其它百分數。該方法不依賴於任何特定rna序列的知識,其中一個優點是鑑定未知的rna-rna相互作用。
只有約5%的基因組編碼翻譯成蛋白質的rna。約50%的基因組轉錄成rna,包括非編碼rna(ncrna)如微小rna和長ncrna(長於200nt)。ncrna通常通過蛋白質相關的相互作用與其他rna相互作用。因此,可以使用基於蛋白質的捕獲方法鑑定直接rna-rna相互作用。在一些實施方案中,可以使用基於蛋白質的捕獲方法鑑定直接rna-rna相互作用。
儘管rna-rna相互作用對於rna的調控功能是至關重要的,但是目前還沒有技術來對其進行全面調查。包括hits-clip(nature460,497-486)和clash(cell153,654-665)的可用技術僅能夠映射所選擇的蛋白質附著的rna。這種一次一種蛋白的方法不能映射整個rna相互作用物組。
在一些實施方案中,本發明的方法和組合物在一次測定中基本上映射了所有蛋白質輔助的rna-rna相互作用。在本文描述的一些實施方案中,已經應用了該方法來產生rna相互作用物組的第一幅全局圖。在一些實施方案中,本發明的方法和組合物規避了對於蛋白質特異性抗體的需求或表達標記蛋白質的需要。這允許rna相互作用物組的無偏倚映射。據我們所知,其他方法一次只能對一種rna結合蛋白起作用。本文描述的實施方案導致了可以為多個rna結合蛋白確定rna-rna相互作用的出乎意料的結果。
在一些實施方案中,本發明的方法和組合物分析內源性細胞狀況,在交聯之前不引入任何外源核苷酸或蛋白質編碼基因(clash)。不需要轉化的細胞系(clash),一些實施方案通常可用於分析任何細胞類型或組織。
在一些實施方案中,本發明的方法和組合物克服了hits-clip的重要缺點。在分析的細胞中,hits-clip推測的rna-rna相互作用並不一定發生。這是因為在hits-clip中共同出現的任意兩種rna可能是由於rna獨立地附著到靶蛋白的不同拷貝上而產生的。然而,在一些實施方案中,本發明的方法和組合物可靠地表示rna的物理相互作用。
已經對小鼠胚胎幹細胞(es)細胞中的rna相互作用物組進行了映射,並且新的發現顯示:
1.長rna經常彼此相互作用。在小鼠es細胞中有數千個mrna-mrna相互作用和數百個lincrna-mrna、轉座子rna-mrna、假基因rna-mrna相互作用。
2.長rna之間的相互作用經常使用一小部分轉錄物。類似於蛋白質相互作用域,本文提出了rna相互作用位點的概念。rna相互作用位點利用鹼基配對來促進長rna的相互作用,這表明了一種新型的反式調控(transregulatory)序列。這些反式調控序列比轉錄物的其他部分在進化上更加保守。
3.rna相互作用物組是一個無標度的網絡,含有幾個高度連接的lincrna和mrna中樞。在一個示例性實施方案中,使用雙色單分子rna-fish已經實驗證實了兩個中樞,malat1lincrna和slc2a3mrna之間的相互作用。
4.基本上每個表達的snorna被酶促加工成mirna樣小rna並與risc複合體中的mrna相互作用。
雖然本發明的方法和組合物的一些實施方案可用於映射分子間相互作用,但是它們也可以揭示關於rna結構的獨特信息。rnahi-c的分子內讀段提供了rna的各個片段的空間鄰近信息。因此,這是第一次以高通量的方式獲得這些信息。另外,在相同的測定中作為副產物獲得每個rna的單鏈區域。在一個示例性實施方案中,rna被蛋白質彎曲,並且通過rnahi-c的分子內讀段捕獲這種四級結構。
在一些實施方案中,所述方法包括:(1)將rna1和rna2與蛋白質(或蛋白質中間體或蛋白質複合體)交聯以形成複合體,(2)標記蛋白質(例如生物素),(3)片段化rna,(4)捕獲標記的蛋白質(例如生物素-鏈黴親和素-珠),(5)將生物素標記的rna接頭連接至rna1和rna2的5'末端,(6)進行鄰位連接以連接rna1-接頭-rna2形成嵌合體,(7)蛋白酶處理複合體以釋放rna1-接頭-rna2嵌合體(dnase處理),(8)與和生物素標記的rna接頭互補的dna探針雜交並用t7外切核酸酶處理以去除非連接的生物素標記的rna接頭,(9)將核酸片段化至約150nt以輔助最終測序,(10)使用鏈黴親和素珠捕獲rna1-接頭-rna2嵌合體,(11)將rna1-接頭-rna2轉化為cdna並測序至少一部分cdna。在一些實施方案中,使用生物信息學來鑑定rna1和rna2。
本發明的方法和組合物可以在多種情況下得到應用,包括rna治療公司尋找新的治療靶點的用途,研究人員研究rna-rna相互作用的用途,以及設備和試劑公司研究和發現設備的開發。
非編碼rna(ncrna)參與包括調控基因表達在內的廣泛細胞過程。微小rna(mirna)和長ncrna(lncrna)是具有已知調控功能的兩類ncrna。這些ncrna在轉錄後或表觀遺傳學水平調控基因表達的能力為基於ncrna的治療提供了新的機會。鑑定ncrna和信使rna(mrna)之間的直接相互作用是了解ncrna調控作用的必然步驟。mirna和lincrna靶點僅是可以通過本文實施方案中描述的技術檢測的相互作用的一小部分,還設計發現了其他ncrna的潛在調控功能。然而,僅由這兩類ncrna驅動的診斷和治療的市場已經是顯著的。
mirna是充當基因表達的關鍵調控劑的一組非編碼核糖核酸。最近的研究進一步揭示了mirna在疾病,特別是癌症、心血管和神經疾病中的重要性。大規模的克隆工作已經揭示了mirna的豐度和多樣性。人類基因組估計可以編碼多達1000種mirna,預計這些mirna可調控所有基因的三分之一。在神經學過程中,mirna是中樞神經系統(cns)發育和可塑性的關鍵調控因子。越來越多的證據表明mirna參與各種各樣的神經病症,如創傷性脊髓損傷、創傷性腦損傷、阿爾茨海默氏病、帕金森病和亨廷頓病。基於mirna的調控的一個有力特徵是單個mirna調控多個功能相關的mrna的能力,如調控多種代謝基因的肝特異性mir-122所例證的。平均來說,給定的mirna可以調控數百個轉錄物,這些轉錄物的效應分子在細胞途徑和網絡內的各個位點起作用。因此,mirna能夠在細胞程序之間瞬時切換,因此通常被視為人類基因組的主要調控因子。
僅在10年前發現了第一種人類mirna,而基於mirna的治療已經進入了第2階段臨床試驗(mir-122拮抗劑,由santaris開發的spc3649,被施用於hcv患者以阻斷病毒複製)。從發現到發展的這種快速進程反映了mirna作為人類疾病中的關鍵調控因子的重要性,並且具有產生一類新的治療藥物的潛力,這可能代表著對目前藥物渠道的有吸引力的補充。
適用於開發基於mirna的治療的原理與採用藥物靶向藥物的路徑的其他靶向治療保持相同。例如,靶標鑑定和驗證是選擇在病因上參與疾病過程的mirna的關鍵。此外,努力的藥物開發對於確保令人滿意的功效、特異性和缺乏毒性是必要的。然而,由於mirna構成與任何其他物質無關的一類藥物靶標,因此還需要新的輔助技術和方法。利用mirna治療潛力的關鍵缺失部分是確定mirna的靶mrna的檢測方法。在一些實施方案中,本發明的方法和組合物可用於開發治療策略和組合物。
癌症治療市場目前接近1000億,預計未來五年將呈指數級增長。基於微小rna的治療已經成為該領域的前沿,根據一些分析師的預計,假設有50種具有治療潛力的mirna被批准使用,基於每個治療性mirna的1.5億美元市場,將佔據價值75億美元的市場空間。
在一些實施方案中,本發明的組合物和方法提供了在任何mirna驅動的治療應用中不能避開的缺失部分。本發明方法和組合物的其它應用包括神經病症中的治療應用和實驗室研究。
lincrna是長於200nt的非蛋白編碼轉錄物,其可以介導表觀遺傳重塑複合體和染色質之間的相互作用。更深入地了解人類癌症中的lncrna功能不僅可以擴展潛在的目標癌症基因的數量,而且還可以促進新型抗癌療法的發展,例如由反義rna介導的基因調控或靶向lncrna-蛋白質相互作用。隨著更深入了解lncrna在正常和疾病狀態中的作用,相信lncrna也可以用作診斷或預測生物標誌物。例如,lncrnahotair在原發性乳腺腫瘤和轉移中的表達增加,其在原發性腫瘤中的表達水平是最終轉移和死亡的有力預測因子。更接近臨床地,在尿液中恰好發現了一種在前列腺癌中高度過表達的稱為前列腺癌抗原3(pca3)的lncrna,使得易於檢測。被稱為progensapca3檢測的商業試劑盒已被fda最近批准用於臨床應用,這是第一個基於尿液的分子檢測來幫助確定重複前列腺活組織檢查的需要。lncrna的疾病調控重要性不僅限於癌症。gibb指出,它們在遺傳性疾病中也發揮重要作用,其中lncrna去調控與短指症和hellp症候群有關。另一種lncrna顯示可穩定阿爾茨海默病途徑中關鍵酶的mrna。越來越多的證據表明,lncrna與主要的人類疾病密切相關,與蛋白質編碼rna相比,可以在疾病診斷和預後方面表現得更好。此外,大多數目前可用的藥物和工具化合物表現出抑制作用機制,並且相對缺乏能夠增加對治療有益的效應子或途徑的活性的藥劑。實際上,在特定情況下,期望許多基因的上調,包括腫瘤抑制子,生長因子,轉錄因子和各種遺傳疾病中缺陷的基因。許多報導表明,lncrna通常可以被rnai觸發子抑制。通過沉默其他基因的rnai來靶向lncrna可以激活基因表達。在一些實施方案中,所述方法和組合物可用於檢測感興趣的細胞中上調基因的存在與否。在一些實施方案中,細胞包含腫瘤細胞、癌細胞或免疫細胞。在一些實施方案中,所述方法可用於通過評估包含上調基因的信息的轉錄組來鑑定或預測疾病或疾病結果。
因此,在一些實施方案中,本發明的方法和組合物可以由使用mirna模擬物來標準化癌細胞上的基因調控網絡、或治療心血管和肌肉疾病的mirna治療市場中的公司使用。在示例性實施例中,本發明的方法和組合物可用於驗證候選產品並且還可以搜索新的目標。
在一些實施方案中,本發明的方法和組合物可用於製備rnahi-c試劑盒。在其它實施方案中,本發明的方法和組合物可用於提供用於研究的寡核苷酸。例如,本發明的方法和組合物可以用於大型lncrna靶向rnai觸發子文庫的環境中。在一些實施方案中,本發明的方法和組合物用於鑑定rnai靶向用的潛在的lncrna候選物。
一個實施方案提供了在細胞中映射出rna-rna相互作用的技術。在一個實施方案中,所述方法和組合物在一個實驗中無偏倚地映射出基本上所有的rna-rna相互作用,並且提供了一對一的解析度(哪個rna與哪個rna相互作用)。一些實施方案包括新的實驗組件和新的計算策略。從某種細胞類型的細胞開始,一些實施方案映射出了該細胞類型的直接相互作用的rna的列表。本發明的方法和組合物已經應用於小鼠胚胎幹細胞,並使用一個實驗鑑定了4049個rna-rna相互作用。在一個實施方案中,實驗組件將這些細胞作為輸入,將基本上所有直接rna-rna相互作用轉化為嵌合rna分子,並使用雙端測序對這些嵌合rna進行序列測定。一些實施方案包括(1)將所有蛋白質-rna複合體(包含蛋白質和核酸的複合體,中間體蛋白質和核酸或蛋白質複合體和核酸)固定在磁珠上;(2)相互作用的rna的基於接近度的連接;(3)嵌合rna分子的選擇性純化;(4)嵌合轉錄物的高通量測序。在本文描述的實施方案中,該方法還可以包括使用生物信息學程序將這些測序數據作為輸入,並產生高置信rna-rna相互作用的列表。
目前,沒有可以直接快速測定細胞類型中基本上所有rna-rna相互作用的有效方法。已有兩種方法來部分實現這一目標,但二者均有缺點。首先,實驗表徵僅一種mirna/lincrna在體內的靶點被認為是一種開創性的技術[lal等人,2011;baigude等人,2012;kretz等人,2013]。其次,可以檢測多種mirna的靶點的其他技術,如hits-clip和clash也有限制。一個主要的常見限制是它們都集中在僅包含小部分rna的mirna。因此,這些技術不能揭示大多數rna-rna相互作用。此外,每種技術都有自己的特殊弱點。
通過交聯免疫沉澱(hits-clip)分離的rna的高通量測序是目前對mirna靶標的全基因組分析的最可靠的方法[chi等人,2009]。hits-clip允許鑑定組織中存在的mirna的總集,以及由mirna調控的mrna的所有總集。然而,直接將mirna與其靶mrna配對不能從hits-clip直接推導出來。換句話說,hits-clip不直接通知哪個mirna調控哪些mrna(沒有一對一的信息)。
最近稱為clash(交聯、連接和混合測序)的方法可以允許直接觀察mirna-靶標對。然而,與測序讀段的數量相比,相互作用的數量仍然很小:只有2%的測序讀段是嵌合的,98%仍然是單一讀段。這需要更深入的測序覆蓋或多個樣品的製備以獲得足夠的mirna-mrna相互作用的覆蓋。
在一些實施方案中,本發明的方法和組合物包括製備和富集rna嵌合體的實驗組件和計算組件,使得可以映射所有rna-rna相互作用的信息的無偏倚的全基因組的直接測定。
在一些實施方案中,本發明的方法和組合物提供:
1.使用嵌合rna以一對一解析度直接測定所有rna-rna相互作用。
2.利用特定的接頭提高連接效率和相互作用鑑別的準確性。
3.選擇性純化期望的嵌合rna-rna產物是通過除去未連接的產物和生物素下拉來實現的。
4.通過使用ssdnacircligase來連接測序適配子而不是rna連接酶,提高用於高通量測序的文庫製備的效率。
在一些實施方案中,本發明的方法和組合物能夠:
1.從實驗步驟產生的所有序列讀段中鑑定嵌合rna序列;
2.將這些嵌合體轉化為注釋的rna簇;
3.使用統計檢驗來確定這些rna簇之間的強直接相互作用。
如前所述,一些技術僅顯示了一種mirna/lincrna在體內的靶點(例如,lal等,2011;baigude等,2012;rna相互作用物組分析)。
如前所述,一些技術可以檢測許多mirna的靶標,但是限於mirna(例如,hits-clip,par-clip,其也缺乏直接的一對一信息,和clash,其僅提供一小部分嵌合rna)。因此,本文描述的本實施方案通過不將rna限制在諸如mirna等小子集而導致相對於現有方法的優點。
在圖4中示出了一個示例性實施方案。簡而言之,細胞通過uv交聯在體內交聯。uv交聯的優點是rna與目的蛋白共價結合,但蛋白質彼此不交聯。rna和蛋白質之間形成的共價相互作用可以使交聯的rna片段得到嚴格的純化。裂解細胞,裂解物經rnasei進行部分rnase消化。此外,在蛋白質上將半胱氨酸殘基生物素化。包括蛋白質-rna複合體(包括蛋白質和核酸的複合體,中間體蛋白質和核酸,或蛋白質複合體和核酸,其中核酸是rna)的蛋白質固定在鏈黴親和素珠上。然後將rna的5'末端與生物素標記的rna接頭(24nt)連接以促進隨後的嵌合rna的選擇性純化。接下來,在有利於交聯rna片段之間連接的稀釋條件下,在珠上進行基於接近度的連接。然後從鏈黴親和素珠洗脫蛋白質-rna複合體(包括蛋白質和核酸的複合體,中間體蛋白質和核酸或蛋白質複合體和核酸,其中核酸是rna),並通過消化結合的蛋白質來回收rna。洗脫的rna進行嚴格的dnase處理以消除dna汙染。然後將純化的rna與同24ntrna接頭互補的dna探針雜交,並用t7外切核酸酶處理以除去非連接的生物素化rna接頭。結果,只有成功連接的嵌合rna在連接點處含有生物素標記的接頭。將該嵌合的rna文庫再次片段化至平均150個核苷酸,並用鏈黴親和素包被的磁珠將連接接合點下拉。終產物是~150nt嵌合rna的文庫。該文庫預期富集r1-接頭-r2形式的嵌合體,其中r1和r2是相互作用的rna的片段。將該文庫轉化為cdna,並用雙端下一代測序進行序列測定。
測序的cdna的生物信息學分析的一個示例性實施方案如圖5所示。首先,對於兩端彼此完全相同的讀段,刪除pcr重複(duplicate)。然後,回收送至測序的片段,並且基於每個讀段對的兩端之間的blast比對來估計片段長度。由此,選擇具有r1-接頭-r2構型的有信息的嵌合rna,其中r1和r2是相互作用的rna的片段(圖5a)。收集嵌合rna後,將r1和r2片段比對回基因組,並且為r1和r2池並行生成由大量重疊的、經比對的讀段支持的簇(使用union-find算法)。
接下來,開展超幾何檢驗以基於連接的嵌合體(r1-接頭-r2)的數量鑑定r1和r2池內的簇之間的強相互作用。不同類型的強相互作用通過r1和r2池中的簇的基因組注釋來確定。(圖5b)
已經進行了使用小鼠胚胎幹(es)細胞的兩個獨立實驗。這兩個實驗產生了可比的結果。cdna的範圍為75至200nt(圖6a,減去128nt引物),其產生~2400萬個非冗餘雙端讀段。鑑定了r1-接頭-r2形式的嵌合rna(240萬個)。通過超幾何檢驗鑑定了總共4049個相互作用,並分類了不同類型的相互作用(圖6b),其中snorna-mrna相互作用最豐富。在242個相互作用中,snorna靶向mrna的3'utr,這支持最近提出的假設,即snorna可以被加工成更小的分子並且像mirna那樣起作用[brameier等人,2011;scott等人,2011]。例如,18個非冗餘嵌合rna將snora1snorna與trim25mrna的3'utr連接(圖6c)。argonaute蛋白質下拉隨後進行rna測序(clip-seq)數據[lueng等人,2011]證實snora1和trim25都與argonaute連接(圖6c)。es細胞分化的時程分析[shu等人,2012]證實了反向相關(圖6d),與一種rna抑制另一種rna的想法一致。
我們的技術的這種原理實驗證據產生了4049對相互作用的rna的列表。基於p值和支持讀段對的數量的前10個相互作用列於表1。
表1:通過rna-stich-seq在胚胎幹細胞中鑑定的前10個rna-rna相互作用。每行提供了一對相互作用的rna的信息,命名為相互作用的rna1和相互作用的rna2。由於這種相互作用對而形成並被反映為雙端測序讀段的嵌合rna的數目列於最後一列。雙向箭頭表示直接相互作用。
許多生物過程受rna-rna相互作用的調控(kretz,m.den等人,controlofsomatictissuedifferentiationbythelongnon-codingrnatincr.nature493,231-235,doi:10.1038/nature11661(2013)),然而分析整個rna相互作用物組仍然是艱巨的。在一個示例性實施方案中,開發了一種方法,rnahi-c,用於在體內映射蛋白質輔助的rna-rna相互作用。通過避免特異性rna結合蛋白的選擇(hafner,m.等人,transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010);chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009);helwak,a.,kudla,g.,dudnakova,t.&tollervey,d.mappingthehumanmirnainteractomebyclashrevealsfrequentnoncanonicalbinding.cell153,654-665,doi:10.1016/j.cell.2013.03.043(2013);kudla,g.,granneman,s.,hahn,d.,beggs,j.d.&tollervey,d.cross-linking,ligation,andsequencingofhybridsrevealsrna-rnainteractionsinyeast.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica108,10010-10015,doi:10.1073/pnas.1017386108(2011)),該方法大大擴展了rna相互作用物組的可鑑定部分。使用這種技術,允許在小鼠胚胎幹細胞中的rna相互作用物組的映射,其由46,780個rna-rna相互作用組成。rna相互作用物組是一個無標度的網絡,其中有幾個作為中樞出現的lincrna和mrna。使用單分子rna螢光原位雜交,在兩個中樞malat1和slc2a3之間驗證了相互作用。在長rna的相互作用位點觀察到鹼基配對,並且該鹼基配對在轉座子rna-mrna和lincrna-mrna相互作用中特別強。這揭示了一種以反式作用的、新型的調控序列。與其假設作用一致,rna相互作用位點比轉錄物的其他區域在進化上更加保守。rnahi-c還提供了關於rna結構的新信息,同時揭示了單鏈區域和每個rna的空間鄰位位點的印跡。因此,具有細胞生理學最小擾動的蛋白質輔助的rna相互作用物組的無偏倚映射對現有方法是有利的,並且將大大擴展調查rna功能的能力。
rna分子之間的相互作用發揮關鍵的調控作用,並且通常由rna結合蛋白(ray,d.等人,acompendiumofrna-bindingmotifsfordecodinggeneregulation.nature499,172-177,doi:10.1038/nature12311(2013))如argonaute蛋白(ago)(meister,g.argonauteproteins:functionalinsightsandemergingroles.naturereviews.genetics14,447-459,doi:10.1038/nrg3462(2013))、pum2、qki(hafner,m.等人,transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010)),和snornp蛋白(granneman,s.,kudla,g.,petfalski,e.&tollervey,d.identificationofproteinbindingsitesonu3snornaandpre-rrnabyuvcross-linkingandhigh-throughputanalysisofcdnas.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica106,9613-9618,doi:10.1073/pnas.0901997106(2009))介導。儘管最近有進展,如par-clip(hafner,m.等人,transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010)),hits-clip(chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009)),和clash(helwak,a.,kudla,g.,dudnakova,t.&tollervey,d.mappingthehumanmirnainteractomebyclashrevealsfrequentnoncanonicalbinding.cell153,654-665,doi:10.1016/j.cell.2013.03.043(2013);kudla,g.,granneman,s.,hahn,d.,beggs,j.d.&tollervey,d.cross-linking,ligation,andsequencingofhybridsrevealsrna-rnainteractionsinyeast.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica108,10010-10015,doi:10.1073/pnas.1017386108(2011)),然而映射所有蛋白質輔助的rna-rna相互作用仍然是一項艱巨的挑戰。
在這三種方法的每一個中,每個實驗只能分析一種rna結合蛋白介導的相互作用。另外,每個實驗需要蛋白質特異性抗體(hits-clip或par-clip)或轉化細胞系中標記蛋白質的穩定表達(clash)。此外,在hits-clip或par-clip中共同出現的任意兩種rna可能是由於rna獨立地附著到靶蛋白的不同拷貝上而產生的。例如,假設細胞中存在10個ago蛋白,每個蛋白都被不同的rna結合;這10個rna將被鑑定為與agohits-clip相互作用。因此,hits-clip和par-clip推斷的rna-rna相互作用在分析的細胞中不一定發生。
在本文所述的示例性實施方案中,開發了rnahi-c方法以檢測體內蛋白質輔助的rna-rna相互作用。在該過程中,rna與其結合的蛋白質交聯,然後連接至生物素化的rna接頭,使得rna,rna1和rna2,由形成rna1-接頭-rna2形式的嵌合rna的相同蛋白質共同結合。使用鏈親和素包被的磁珠分離這些含接頭的嵌合rna,並使其進行雙端測序(方法,圖1a,圖7)。因此,每個非冗餘雙端讀段反映一種分子相互作用。
rnahi-c方法提供了映射rna-rna相互作用的幾個優點。首先,僅捕獲由相同蛋白質分子聚集的rna,克服了hits-clip中的缺點,在hits-clip中當不同的rna獨立地結合到相同蛋白質的不同拷貝時,其將所述不同的rna視為是相互作用的。第二,使用生物素化的接頭作為選擇標誌物避免對於蛋白質特異性抗體的需求或表達帶標記的蛋白的需要。這允許rna相互作用物組的無偏倚映射。如本領域所述,其它方法一次只能對一種rna結合蛋白起作用。因此,這種方法導致一次有效地對多於一種rna結合蛋白起作用的驚人效果。第三,通過在極度稀釋的條件下在鏈黴親和素珠上進行rna連接步驟,使得由rna隨機連接至其他附近rna產生的假陽性最小化。第四,rna接頭提供了一個清晰的邊界,描繪跨越連接位點的測序讀段,從而避免了映射測序讀段的含糊不清。第五,rnahi-c直接分析內源細胞條件而在交聯之前不引入任何外源核苷酸(hafner,m.等人,transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010);lal,a.deng等人,captureofmicrorna-boundmrnasidentifiesthetumorsuppressormir-34aasaregulatorofgrowthfactorsignaling.plosgenetics7,e1002363,doi:10.1371/journal.pgen.1002363(2011);baigude,h.,ahsanullah,li,z.,zhou,y.&rana,t.m.mir-trap:abenchtopchemicalbiologystrategytoidentifymicrornatargets.angewchemintedengl51,5880-5883,doi:10.1002/anie.201201512(2012))或蛋白結合基因(helwak,a.,kudla,g.,dudnakova,t.&tollervey,d.mappingthehumanmirnainteractomebyclashrevealsfrequentnoncanonicalbinding.cell153,654-665,doi:10.1016/j.cell.2013.03.043(2013))。第六,通過在pcr擴增之前將隨機的6個核苷酸條形碼附著至各嵌合rna並隨後僅一次計數與相同的條形碼完全重疊的測序讀段來除去潛在的pcr擴增偏倚(chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009);loeb,g.b.deng’ren等人,transcriptome-widemir-155bindingmaprevealswidespreadnoncanonicalmicrornatargeting.molecularcell48,760-770,doi:10.1016/j.molcel.2012.10.002(2012);wang,z.等人,iclippredictsthedualsplicingeffectsoftia-rnainteractions.plosbiology8,e1000530,doi:10.1371/journal.pbio.1000530(2010);konig,j.等人,icliprevealsthefunctionofhnrnpparticlesinsplicingatindividualnucleotideresolution.naturestructural&molecularbiology17,909-915,doi:10.1038/nsmb.1838(2010))。
在一個示例性實施方案中,以較小的技術差異對小鼠胚胎幹(es)細胞進行了兩個獨立的rnahi-c測定(圖8-12),其被稱為es-1和es-2。為了控制由大蛋白質複合體組裝的rna(zhao,j.等人genome-wideidentificationofpolycomb-associatedrnasbyrip-seq.molecularcell40,939-953,doi:10.1016/j.molcel.2010.12.011(2010))或細胞器而不是單個蛋白質,使用在核苷酸和蛋白質之間以及在蛋白質之間形成共價鍵的兩個交聯劑(甲醛和egs)產生rnahi-c文庫(es間接)(nowak,d.e.,tian,b.&brasier,a.r.two-stepcross-linkingmethodforidentificationofnf-kappabgenenetworkbychromatinimmunoprecipitation.biotechniques39,715-725(2005);zeng,p.y.,vakoc,c.r.,chen,z.c.,blobel,g.a.&berger,s.l.invivodualcross-linkingforidentificationofindirectdna-associatedproteinsbychromatinimmunoprecipitation.biotechniques41,694,696,698(2006))。另一個文庫由小鼠胚胎成纖維細胞(mef)產生,為生物信息學質量評估提供了又一個數據集(圖13)。證實每個文庫含有期望形式(rna1-接頭-rna2)和長度的rna構建體(圖1b)。對每個文庫測序,平均得到4730萬個雙端讀段,其中大約1510萬個非冗餘雙端讀段代表期望的嵌合形式(圖1c)。
創建了一組生物信息學工具(rna-hic-工具)來分析和顯現rnahi-c數據(圖14-15)。rna-hic-工具自動化分析步驟,包括去除pcr重複,分離復用樣品,鑑定接頭序列,分離連接點讀段,召集(call)相互作用rna,進行統計學評估,分類rna相互作用類型,召集相互作用位點和分析rna結構(方法)。它還為rna相互作用物組和rna內的鄰位位點提供可視化工具(圖16)。
比較四種rnahi-c文庫。通過fpkms的相關性(針對接頭的左側和右側的讀段片段分別計算)判斷es-1和es-2最相似,接著是es-間接,然後是mef(圖13)。從es-1和es-2鑑定的相互作用的rna對表現出強重疊(p值<10-35,置換檢驗)。在mef中鑑定的相互作用與es樣品中的那些沒有顯著的重疊(每個重疊的p值=1,置換檢驗)。例如,trim25rna的3'utr與小核仁rna(snorna)snora1之間的相互作用分別由es-1和es-2樣品中的24個和22個雙端讀段支持,但在es-間接或mef文庫中未檢測到(圖1c)。包括snora1在內的被鑑定為與mrna相互作用的多達172個snorna由agohits-clip(圖1c)和小rna測序數據支持(u,p.etal.spatiotemporalclusteringoftheepigenomerevealsrulesofdynamicgeneregulation.genomeresearch23,352-364,doi:10.1101/gr.144949.112(2013))(圖1c,圖17-19),表明大多數表達的snorna基因被酶促加工成mirna樣小rna並與risc複合體中的mrna相互作用(ender,c.等人,ahumansnornawithmicrorna-likefunctions.molecularcell32,519-528,doi:10.1016/j.molcel.2008.10.017(2008);brameier,m.,herwig,a.,reinhardt,r.,walter,l.&gruber,j.humanboxc/dsnornaswithmirnalikefunctions:expandingtherangeofregulatoryrnas.nucleicacidsres39,675-686,doi:10.1093/nar/gkq776(2011))(文本s1)。
然後需要知道其他rna是否可以經歷與mirna生物發生相似的過程,並且也與mrna相互作用。為此,rnahi-c鑑定的相互作用的rna與通過小rna測序(小rna-seq)發現的那些和與es細胞中的ago蛋白(hits-clip)結合的那些相交(s.w.chi,j.b.zang,a.mele,r.b.darnell,argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479(jul23,2009))。小rna-seq選擇性測序,「mirna和具有由dicer或其他rna加工酶的酶促裂解產生的3'羥基的其他小rna」(illumina,「truseq(r)samllrnasamplepreparationguide」(2014))。除了mirna之外,包括snorna、假基因rna、mrnautr等其他rna類型也有貢獻於小rna池,並附著於ago(圖17a)。此外,大部分rnahi-c鑑定的相互作用的rna對共同出現在agohits-clip數據中(圖18)。該數據表明存在由dicer或其他rna加工酶消化並被併入risc複合體的非mirna。
為了闡明哪些類型的非mirna基因最可能經歷mirna樣生物發生,rnahi-c鑑定的rna-rna相互作用經受以下過濾:
1.相互作用涉及一個mrna(稱為靶)和一個其他rna(源rna);
2.通過酶裂解將源rna加工成小rna(小rna-seq中的fpkm>0);
3.靶rna和源rna都出現在agohits-clip(兩個rna的fpkm>0);
4.源rna和靶rna的rnahi-c鑑定的相互作用位點表現出強的鹼基配對(p值<0.05,wilcoxon符號秩檢驗,比較每一雙端讀段的rna1和rna2序列之間的結合能與隨機改組的核苷酸序列的結合能)。
總共302個rna-rna相互作用通過了這些過濾。這些相互作用中大多數(79%)的源rna是snorna(表2)。因此,snorna優先進行功能分析。
表2.mirna樣rna。通過(1)涉及mrna(稱為靶)和另一個rna(稱為源rna),(2)源rna存在於小rna-seq中,(3)靶rna和源rna出現在agohits-clip中,(4)源rna和靶rna的rnahi-c鑑定的相互作用位點表現出強的鹼基配對,過濾rnahi-c鑑定的rna-rna相互作用。第2列列出了滿足標準1-3的相互作用位點的數量。第3列列出了滿足標準1-4的相互作用位點的數量。第4列列出了滿足標準1-4的相互作用位點的數量。
假設大量snorna被酶促加工成mirna樣短rna並與mrna相互作用。這一假設得到了919個rnahi-c鑑定的snorna-mrna相互作用的支持,其中mrna和snorna都被ago結合。此外,ago結合的snorna及其相互作用的mrna在es細胞向中內胚層的指導分化期間顯示出反相關表達變化(p.yu等人,spatiotemporalclusteringoftheepigenomerevealsrulesofdynamicgeneregulation.genomeresearch23,352(feb,2013))(圖17b)。另外,與沒有ago結合的相比,ago結合的snorna及其靶mrna表現出更強的鹼基配對(圖17c)。最後,用作參考地,從snorna處理的小rna與mrna的utr區域相互作用。在涉及rna-rna相互作用的497個snorna中,243個與utr區域相互作用,其中在小rna-seq中檢測到223個(92%),表明經歷了酶切(圖17d)。相比之下,與非utr區相互作用的其他254個snorna含有較少的(55%)小rna。此外,相比於與非utr相互作用的snorna,兩倍以上的utro相互作用的sno-sirna是ago結合的(p值<2.2-16,卡方檢驗)。例如,snora14rna靶向mcl1mrna的3'utr(圖19a)。snora14rna(110-135nt)上的相互作用位點與酶處理的小rna以及ago結合區精確重疊。snora14rna的酶促處理部分完全位於髮夾環的一側(圖19b),並且對mcl1utr上的靶位點顯示出強結合親和力(-60kcal/mol)。經處理的snora14rna的表達與mcl1mrna的表達反相關(圖19c)。總之,該數據表明大量小幹擾rna源自snorna基因,其與es細胞中的900個以上的mrna相互作用。
合併es-1和es-2文庫以推斷es細胞中的rna相互作用物組。這些數據包括454萬個非重複的雙端讀段,將其明確地分成兩個rna片段,兩個片段都獨特地映射到基因組(mm9)。鑑定了46,780個rna間相互作用(fdr<0.05,fisher精確檢驗)(圖20)。mrna-snorna相互作用是最豐富的類型,儘管還檢測到數千個mrna-mrna和數百個lincrna-mrna,假基因rna-mrna,mirna-mrna相互作用(圖21)。這可能是任何生物體中描述的第一個rna相互作用物組。因此,對於整個實驗和分析程序該模擬表明了大約66%的靈敏度和93%的特異性(文本s2)。
rnahi-c的模擬分析
1.1數據合成。為了評估rnahi-c的靈敏度和特異性(包括其實驗和計算程序),進行了模擬分析。模擬了通過計算模擬數據生成過程的1,000,000個雙端讀段。用於模擬的參數是從實際數據中得出的。模擬數據生成過程如下。
對於每個雙端讀段(2×100個鹼基):
1.選擇來自具有相同概率的四個樣品條形碼的樣品條形碼,並將其與6nt隨機條形碼串聯(如圖15a所示)。
2.分別以[0.1、0.3、0.1、0.3、0.2]概率將該雙端讀段分配到[僅接頭、無接頭、rna1-接頭、接頭-rna2、rna1-接頭-rna2]的列表中的一種類型的cdna(如圖15c所示)。
3.如果將此讀段對分配給含接頭的類型,則以相等的概率隨機選擇1或2個接頭。應注意,少量的含接頭的讀段對包含2個接頭;使用相等的概率是估計最壞情況的保守選擇。
4.根據步驟2中確定的cdna類型,產生rna1和rna2部分的序列。對於rna1和rna2兩者,
a.由l~unif(15,150)模擬其長度,
b.基於以下概率從[「mirna」、「mrna、「lincrna」、「snorna」、「snrna」、「trna」]中選擇rna類型:
i.如果長度l<50,使用[0.2、0.2、0.1、0.2、0.2、0.1],
ii.否則,使用[0.05、0.4、0.2、0.2、0.1、0.05];
c.根據ensembl採集的rna型(釋放67,小鼠ncbim37)隨機選擇一種rna,
d.從所選rna隨機取長度l的序列片段。
5.串聯由步驟1、3、4生成的條形碼、接頭和rna片段,產生合成cdna序列。
6.如果步驟5中的合成cdna為100bp以上,則分別從正義鏈和反義鏈的合成cdna的兩端取100個鹼基。
7.如果步驟5中的合成cdna短於100bp,則將其正義鏈和反義鏈分配為正向讀段和反向讀段,並將p5和p7引物序列連接至兩個讀段。
8.在每個鹼基上以0.01的比率模擬測序誤差(n.j.loman等人,performancecomparisonofbenchtophigh-throughputsequencingplatforms.naturebiotechnology30,434(may,2012))。
步驟1-5根據實驗程序模擬了cdna序列,步驟6-8基於該cdna序列模擬了雙端讀段。保持模擬的相互作用的rna對,以及每個部分(rna1、接頭和rna2,如果適用)的cdna類型和長度,用於與計算預測進行比較。
1.2.評估中間結果和最終結果。合成數據用於評估兩個中間分析步驟以及最終預測的靈敏度和特異性。
首先,比較預測的cdna長度(rna-hic-工具的步驟3的輸出)與實際長度(表3)。該步驟「3.在測序文庫中回收cdna」將每個cdna根據它們的長度分配為四種類型,即類型1(200bp);類型4(未知)(圖32)。該算法實現了對每種類型的預測的高靈敏度和特異性。只有很少(0.58%)的短於200bp的cdna預測為超過200bp。這些錯誤是由於正向和反向讀段的小重疊(通常在0到5bps之間),這無法由局部比對所檢測到。
表3.預測和真實cdna長度範圍的比較。將每種類型的預測cdna(第1-4列)的計數與其真實類型(行)進行比較。
當預測長度短於200bp(類型1和類型2)時,可以預測精確的長度。在這些情況下,預測長度通常與模擬cdna的長度精確匹配(圖33a)。
接下來,比較每個cdna的預測嵌合構型(rna-hic-工具的步驟4的輸出)與合成構型。在步驟「4.解析嵌合cdna」中,基於接頭序列的存在,該算法將cdna分為五類。該算法對「rna1-接頭-rna2」形式的cdna靈敏度達到99.89%,特異性為95.82%(表4)。
表4.預測的和真實的cdna構型的比較。將預測構型(列)的cdna數與其真實構型(行)進行比較。
最後,比較預測的和模擬的rna-rna相互作用。模擬數據集包含200,200個嵌合rna對,其中檢測到131,571對rna(靈敏度=65.72%,特異性=92.57%,圖33c)。還分別計算了每種類型rna的相互作用的靈敏度和特異性(圖33c)。無論參與的rna類型如何,該方法顯示很少的假陽性(特異性≥90%)。不涉及轉座子rna或snrna的相互作用比那些涉及的顯示出更少的假陰性。這是由於轉座子和snrna序列的重複性質。最嚴重的情況涉及linerna,其中靈敏度下降到52%。據保守估計,涉及轉座子rna的相互作用約有一半可能被該程序錯過。估計約2/3至3/4的不涉及轉座子rna的相互作用將被鑑定出來。
每個rna的相互作用的伴侶的數量是非常不平衡的。es細胞rna相互作用物組是無標度網絡,其度分布符合冪定律(p(k)~k-γ,γ=3)(圖22a)(barabasi,a.l.&oltvai,z.n.networkbiology:understandingthecell'sfunctionalorganization.naturereviews.genetics5,101-113,doi:10.1038/nrg1272(2004))。為了查看無標度性質是否由少量高度連接的snorna、snrna和trna驅動,將它們從網絡中移除。僅由mrna、lincrna、mirna、假基因rna和反義rna組成的相互作用保持無標度(圖22b)。許多mrna、假基因rna和lincrnas作為中樞出現(具有大量連接的節點,圖1d)。最大的mrna中樞是suv420h2,其與21個mrna和2個lincrna相互作用。最大的lincrna中樞是malat1,其與4個mrna相互作用,包括slc2a3的mrna中樞。
大多數(83.05%)的相互作用的rna顯示重疊的rnahi-c讀段(圖2a),表明相互作用通常集中在rna的特定區段。鑑定重疊閱讀片段的「峰」,稱為「相互作用位點」(圖2b)。相互作用位點不僅出現在mirna(整個成熟mirna)mrna、lincrna上,而且也出現在假基因和轉座子rna上(圖2c)。在l1、sine、ervk、malr和erv1轉座子rna中存在超過2000個相互作用位點(圖23),表明它們與其他rna頻繁相互作用(shalgi,r.,pilpel,y.&oren,m.repressionoftransposable-elements-amicrornaanti-cancerdefensemechanism?trendsingenetics:tig26,253-259,doi:10.1016/j.tig.2010.03.006(2010);yuan,z.,sun,x.,liu,h.&xie,j.micrornagenesderivedfromrepetitiveelementsandexpandedbysegmentalduplicationeventsinmammaliangenomes.plosone6,e17666,doi:10.1371/journal.pone.0017666(2011))。
推測鹼基互補是否被不同類型的rna-rna相互作用利用。一對相互作用的rna的雜交能通過連接片段對(rna1,rna2)的平均雜交能評估(bellaousov,s.,reuter,j.s.,seetin,m.g.&mathews,d.h.rnastructure:webserversforrnasecondarystructurepredictionandanalysis.nucleicacidsres41,w471-w474,doi:doi10.1093/nar/gkt290(2013)),並與鹼基的隨機改組產生的對照rna的雜交能進行比較。互補鹼基在幾乎所有類型的rna-rna相互作用中都是優選的,並且在轉座子rna-mrna、mrna-mrna、假基因rna-mrna,lincrna-mrna、mirna-mrna相互作用(p值<2.4-18)中最顯著,但是在ltr-假基因rna相互作用中沒有觀察到(圖2d,圖24)。該數據表明一種新的機制,其中鹼基配對有助於長rna中序列特異性的轉錄後調控。
如果這些rna-rna相互作用是序列特異性的,則rna相互作用位點應該處於選擇性壓力之下。發現種間保守水平(cooper,g.m.等人,distributionandintensityofconstraintinmammaliangenomicsequence.genomeresearch15,901-913,doi:10.1101/gr.3577405(2005))在相互作用位點處強烈增加,保守峰精確地定位兩個rna片段的連接點(圖2d)。當與lincrna、假基因rna、轉座子rna或其他mrna相互作用時,mrna上的相互作用位點比其餘轉錄物更保守(圖25)。lincrna和假基因rna上的相互作用位點在lincrnas-mrna、假基因rna-mrna和假基因rna-轉座子rna相互作用中表現出增加的保守性(圖25)。相互作用位點上增加的保守性不是由於外顯子-內含子的邊界(圖26)。總之,鹼基互補在長rna的相互作用中廣泛傳播,並被進化選擇。這表明了基因組中編碼的新型調控信息。
儘管rnahi-c最初設計用於映射分子間相互作用,但是發現rnahi-c揭示了rna二級結構和三級結構。以上分析均基於分子間讀段。通過閱讀分子內讀段,可以了解rna結構的幾個方面。首先,通過rnasei消化位點的密度(連接前應用rnasei消化,參見圖1a中的步驟2,圖27)鑑定rna的單鏈區域的足跡。第二,通過鄰位連接捕獲每個rna的空間鄰位位點(圖1a中的步驟5)。總共67,221個讀段對映射到各個基因,但不在彼此或相同的鏈上的2,000bp內,因此是從分子內切割和連接產生的(圖28a)。通過將測序讀段中的rna1和rna2的取向與其在基因組中的取向進行比較,每個切割和連接的序列可以明確地分配到兩個結構類別之一(圖3a)。例如,從snora73轉錄物產生277個切割和連接的序列(圖3b)。rnasei消化位點的密度(圖3c)強烈地預測了rna的單鏈區域(熱圖,圖3e)。檢測到6對鄰位位點(圓,圖3d)。每對由三個以上的具有重疊連接位置的切割和連接的序列支持(黑點,圖3b)。六個鄰位位點對中的五個在通常接受的二級結構中物理上接近(箭頭,圖3e)。在snora14上,根據測序推斷的二級結構,一對推斷的鄰位位點出現較遠(圖29)。然而,核糖核蛋白dyskerin在體內彎曲snora14轉錄物(kiss,t.,fayet-lebaron,e.&jady,b.e.boxh/acasmallribonucleoproteins.molecularcell37,597-606,doi:10.1016/j.molcel.2010.01.032(2010)),使兩個假尿苷酸化環彼此接近,如通過切割和連接的序列所預測的(圖3f)。結構信息甚至可以在新的轉錄物和mrna的一些部分得到(圖30-31)。迄今為止,解析任何單個rna的空間鄰位鹼基仍然是一項艱巨的挑戰。rnahi-c為數千個rna提供分子內空間鄰位信息。此外,每個rna的單鏈足跡同時映射。因此,rnahi-c大大擴大了我們檢測rna結構的能力。
映射rna相互作用的關鍵是選擇。在rnahi-c中引入可選擇的接頭使得能夠無偏倚地選擇相互作用的rna,使得可以全局地映射rna相互作用物組。es細胞中每個rna的相互作用伴侶的數量是非常不平衡的,導致無標度的rna網絡。長rna之間的相互作用經常使用一小部分轉錄物。類似於蛋白質相互作用域,提出了rna相互作用位點的概念。rna相互作用位點利用鹼基配對來促進長rna的相互作用,提示了一種新型的反式調控序列。這些反式調控序列比轉錄物的其他部分在進化上更加保守。rna結構也可以通過rnahi-c進行映射。本文提供了其中rna被蛋白質彎曲的示例性實施方案,並且通過rnahi-c的分子內讀段顯示這種三級結構。因此,這種方法和數據應該大大有助於將來對rna功能和調控作用的調查。
軟體訪問
rna-hic-工具軟體可在http://systemsbio.ucsd.edu/rna-hi-c獲得,其公開內容通過引用整體併入本文。
材料和方法
細胞培養
未分化小鼠e14es細胞在無飼養條件下培養。將es細胞接種在明膠包被的培養皿上,並在補充有15%胎牛血清(fbs;geminigemcell)、0.055mm2-巰基乙醇(sigma)、2mmglutamax(gibco)、0.1mmmem非必需胺基酸(gibco),5,000u/ml青黴素/鏈黴素(gibco)和1,000u/mllif(millipore)的杜氏改良的eagle培養基(dmem;gibco))中培養。將細胞保持在37℃和5%co2的培養箱中。
在15cm培養皿中在補充有15%胎牛血清(fbs;geminigemcell)、0.055mm2-巰基乙醇(sigma)、2mmglutamax(gibco)、0.1mmmem非必需胺基酸(gibco)、5,000u/ml青黴素/鏈黴素(gibco)的dmem(gibco)中培養小鼠胚胎成纖維細胞(mef)。mef也保持在37℃和5%co2的培養箱中。
將果蠅s2細胞(invitrogen)保持在28℃的無co2培養箱中的15cm平板中的補充有10%熱滅活的胎牛血清(fbs;geminigemcell)和5ml1:100青黴素-鏈黴素的施耐德果蠅培養基(schneider'sdrosophilamedium,gibco)中。
組織解剖和準備
小鼠處理由加利福尼亞大學聖地牙哥分校的機構動物護理和使用委員會批准。成年雌性(c57bl/6j背景)通過頸脫位處死並立即收集全腦,用冰冷pbs衝洗三次並快速冷凍。使用研缽和研杵將冷凍的全鼠腦組織在液氮中研磨成細粉末。將組織粉末快速轉移到乾冰床上的陪替氏培養皿中,並在uv交聯劑(254nm)中以400mj/cm2在乾冰上照射三次,每次照射之間輕輕渦旋。立即將交聯的粉狀組織溶解並進行如所述的rnahi-c程序。
rnahi-c方法概述
rnahi-c設計為:(i)以無偏倚方式在體內捕獲相互作用的rna,而不遺傳地或瞬時地引入外源分子;(ii)允許嚴格去除細胞裂解後形成的非生理關聯(s.mili,j.a.steitz,rna10,1692(2004));(iii)選擇鄰位連接的嵌合rna;(iv)允許相互作用的rna的明確的生物信息學鑑定。這些目標可以通過以下方式實現:(i)所有rna-蛋白質複合體(包括蛋白質和核酸的複合體,含核酸的中間體蛋白質或結合至核酸的蛋白質複合體,其中核酸為rna)在鏈黴親和素珠中的交聯和固定,並通過變性條件去除非特異性結合;(ii)連接生物素標記的rna接頭以促進嵌合rna構建體的選擇性富集;(iii)使用接頭序列從測序讀段對中明確分離相互作用的rna。
步驟1:將rna與蛋白質交聯
uv照射用於在光活性核苷酸鹼基和胺基酸之間形成共價鍵。uv照射在rna內產生高反應性、短壽命的狀態的核苷酸鹼基,僅在其接觸點與胺基酸形成共價鍵,而沒有可能引起構象擾動的附加元件(i.g.pashev,s.i.dimitrov,d.angelov,trendsinbiochemicalsciences16,323(1991))。254nm處的uv照射不會由於胺基酸吸收的不同波長而促進蛋白質-蛋白質的交聯。具體地,將細胞在冰冷的pbs中洗滌兩次,並在冰上冰冷的pbs中以400mj/cm2的uv-c(254nm)照射。通過刮擦收穫細胞,並在4℃下以1,000×g離心5分鐘來沉澱細胞。細胞沉澱物在液氮中快速冷凍並儲存在-80℃。
產生rnahi-c文庫(es間接),其中蛋白質-蛋白質複合體也是交聯的。這是為了捕獲通過蛋白質相互作用匯集在一起的rna。以先前驗證的參數實施體內雙重交聯法(illumina,「truseq(r)samllrnasamplepreparationguide」(2014);p.yu等人,spatiotemporalclusteringoftheepigenomerevealsrulesofdynamicgeneregulation.genomeresearch23,352(feb,2013);n.j.loman等人,performancecomparisonofbenchtophigh-throughputsequencingplatforms.naturebiotechnology30,434(may,2012))。簡言之,首先用室溫pbs衝洗細胞,並在室溫下在搖床上用新鮮製備的pbs中的1.5mm乙基甘醇二(琥珀醯亞胺基琥珀酸酯)(egs,pierceproteinresearchproducts,rockford,illinois)處理細胞45分鐘。用甲醛(pierceproteinresearchproducts,rockford,illinois)進一步處理細胞至終濃度為1%,並在室溫下搖動孵育20分鐘。加入甘氨酸至終濃度為250mm,並在室溫下孵育10分鐘以淬滅交聯反應。然後將細胞用pbs在室溫下洗滌一次,刮掉,在4℃下以1,000×g沉澱5分鐘,在液氮中快速冷凍並儲存在-80℃。
進行對照實驗(es間接),其中蛋白質-蛋白質複合體也是交聯的。這為通過蛋白質相互作用匯集在一起的rna提供對照。因此,以先前驗證的參數實施體內雙重交聯法(s.k.kurdistani,m.grunstein,methods31,90(2003);d.e.nowak,b.tian,a.r.brasier,biotechniques39,715(2005);j.zhang等人,methods58,289(2012))。簡言之,首先用室溫pbs衝洗細胞,並在室溫下在搖床上用新鮮製備的pbs中的1.5mm乙基甘醇二(琥珀醯亞胺基琥珀酸酯)(egs,pierceproteinresearchproducts,rockford,illinois)處理細胞45分鐘。用甲醛(pierceproteinresearchproducts,rockford,illinois)進一步處理細胞至終濃度為1%,並在室溫下搖動孵育20分鐘。加入甘氨酸至終濃度為250mm,並在室溫下孵育10分鐘以淬滅交聯反應。然後將細胞用pbs在室溫下洗滌一次,刮掉,在4℃下以1,000×g沉澱5分鐘,在液氮中快速冷凍並儲存在-80℃。
步驟2:細胞裂解、rna片段化和蛋白質生物素化
將儲存在-80℃的大約6×108個交聯的細胞在冰上融化並重懸於~3體積的裂解緩衝液(50mmtris-hclph7.5,100mmnacl,0.1%sds,1%igepalca-630,0.5%脫氧膽酸鈉,1mmedta,補充有1:20體積的無edta完全蛋白酶抑制劑混合物(roche))。在冰上進行裂解20分鐘。通過在4℃下以20,000×g離心10分鐘除去細胞碎片和不溶性染色質。收集上清液,並用濃度為10μlturbodnase/ml裂解物的turbodnase(invitrogen)在37℃處理20分鐘。通過每ml裂解物加入10μl的1:100稀釋的rnasei(neb)並在37℃下孵育3分鐘,將rna消化成~1000-2000nt(es-1)或~1000nt(es-2)片段。在rnasei處理後,立即將裂解物轉移至冰中至少5分鐘。rnasei和基於超聲處理的片段化均留下與rna連接不相容的5'-oh和3'-p末端,抑制不需要的rna連接。為了停止dnase消化,添加edta(ambion)至25mm終濃度,並在4℃下旋轉孵育混合物15分鐘。片段化的雙重交聯(es間接)裂解物如下製備:在冰上裂解20分鐘後,懸浮液通過在下述設置下的超聲處理(covarise220)直接進行片段化:4℃下,20分鐘,具有5%的佔空比,峰值入射功率為140瓦,每次爆發(burst)200個循環。
對於跨物種實驗(fly-mm),分別裂解約3×108個e14mes細胞和3×108個果蠅s2細胞,然後在蛋白質生物素化之前將其混合。
為了解離鬆散結合的蛋白質,加入終濃度500mm的nacl,並將溶液在4℃下旋轉孵育10分鐘。為了進一步解離蛋白複合體和非交聯rna並停止rnasei的活性,加入sds至終濃度0.3%,並在65℃下在750r.p.m下振蕩孵育混合物15分鐘。使溶液混合物冷卻至室溫後,通過加入裂解物1:5體積的25mm(13.56mg/ml)ezlink碘乙醯基-peg2-生物素(ipb)(pierceproteinresearchproducts)並在室溫下將混合物在黑暗中旋轉90分鐘來使半胱氨酸殘基生物素化。通過加入dtt至5mm濃度並在室溫下孵育15分鐘,淬滅生物素化反應。為了中和sds,加入tritonx-100(sigma)至2%終濃度,並在37℃下孵育15分鐘。將裂解物樣品在20kd截留的slide-a-lyzer透析盒(pierceproteinresearchproducts,rockford,illinois)中在2升透析緩衝液(20mmtris-hclph7.5,1mmedta)中室溫下透析以去除過量的生物素。透析緩衝液至少更換三次,每2小時一次。透析後,將裂解物轉移到15ml管中。
步驟3:珠上固定化
蛋白質-rna複合體以低珠表面密度固定在鏈黴親和素包被的珠上(800μlmyone鏈黴親和素t1珠,相當於200cm2表面積)。在固體表面上固定化的優點包括:(i)減少非交聯寡核苷酸之間的隨機分子間連接(r.kalhor,h.tjong,n.jayathilaka,f.alber,l.chen,natbiotech30,90(2012)),(ii)允許有效的緩衝液交換,(iii)通過嚴格洗滌去除非生理相互作用。
800μlmyonet1珠用pbst(含有0.1%tween-20的pbs)洗滌三次,重新懸浮於800μl相同的緩衝液中並轉移到生物素化的裂解物中。將珠-裂解物懸浮液在室溫下旋轉45分鐘。在該孵育期間,通過加入相等摩爾數的dtt並在室溫下孵育至少30分鐘來製備200μl中和的25mmipb。使用磁性支架將珠固定,並將大部分上清液吸出,留下4ml上清液。將珠在剩餘的溶液中重懸,然後加入200μl中和的ipb。ipb用於將固定化後過量的未結合的鏈黴親和素飽和,過量的未結合的鏈黴親和素可幹擾涉及生物素標記的rna接頭的後續步驟。為了除去非共價連接至蛋白質或經非特異性蛋白質-蛋白質相互作用的不需要的rna(ss.c.kwon等人,natstructmolbiol20,1122(2013);a.castelloetal.,nat.protocols8,491(2013)),4℃下將珠用冰冷的變性洗滌緩衝液i(50mmtris-hclph7.5,0.5%十二烷基硫酸鋰,500mm氯化鋰,7mmedta,3mmegta,5mmdtt)旋轉洗滌三次,每次5分鐘。然後將珠用冰冷的高鹽洗滌緩衝液ii(50mmtris-hclph7.5,1mnacl,0.1%sds,1%igepalca-630,1%脫氧膽酸鈉,5mmedta,2.5mmegta,5mmdtt),洗滌緩衝液iii(1×pbs,1%tritonx-100,1mmedta,1mmdtt)和pnk洗滌緩衝液(20mmtris-hclph7.5,10mmmgcl2,0.2%tween-20,1mmdtt)洗滌;各緩衝液在第二次洗滌期間在4℃旋轉5分鐘兩次。
步驟4:生物素-標記的rna接頭的連接
接下來,將生物素標記的rna接頭(5'-rcrurarg/ibiodt/rargrcrcrcrarurgrcrararurgrcrgrargrgra)(seqidno:1)連接至rna的5'末端。生物素標記的接頭充當用於富集連接的rna的選擇標記;其也勾畫出明確的邊界,以明確地分離覆蓋連接接合點的任何測序讀段。rna接頭的5'末端暫時「阻止」連接,以避免接頭環化或串聯。這是通過合成含5'-oh基團的接頭而實現的,該基團與連接不相容但可以通過磷酸化被「再活化」。然而,rnasei留下與接頭連接不相容的5'-oh末端,因此5'末端首先用t4多核苷酸激酶(pnk),3'磷酸酶減(neb)進行磷酸化。野生型t4pnk由於其額外的3'磷酸酶活性而無法使用,其額外的3'磷酸酶活性將rna的3'末端從3'-p修飾為3'-oh,使其易於自連接。
這通過去除洗滌緩衝液並隨後將珠重懸於100μlpnk反應混合物(73μl無rnase的水,10μl10×pnk緩衝液,10μl10mmatp,5μl10u/μlt4pnk(3'磷酸酶減)(neb),2μlrnasinplus(promega)),並在37℃下孵育1小時,每2分鐘在1,200rpm下間歇振蕩5秒來實現。用洗滌緩衝液i、ii、iii和pnk洗滌珠,每次緩衝液在第二次洗滌中在4℃旋轉5分鐘兩次。使用冰冷的洗滌液來消除任何可磷酸化rna接頭誘導其可能連接至rna的3'末端的剩餘的pnk。洗滌緩衝液除去後,通過加入含有2μlrnasinplus(promega),16μl10mmatp,16μl10×rna連接酶緩衝液,16μl1mg/mlbsa,30μl20μm生物素標記的接頭,64μl50%peg8000(neb),16μl10u/μlt4rna連接酶1(neb)的160μlrna連接反應混合物,將生物素標記的rna接頭連接至rna5'末端。連接在37℃下進行1小時,在16℃下進行過夜,每2分鐘在1,200r.p.m下間歇振蕩15秒。加入bsa以增強t4rna連接酶的活性並防止珠聚集。peg用於通過增加供體和受體末端的濃度來增強分子間連接(d.b.munafó,g.b.robb,rna16,2537(2010))。
步驟5:鄰位連接
接下來,將珠用冰冷的洗滌緩衝液ii洗滌兩次,用冰冷的洗滌緩衝液iii和pnk洗滌緩衝液洗滌一次。為了準備鄰位連接,首先使用t4pnk的3'磷酸酶活性對rna3'-末端進行脫磷酸化,留下3'-羥基(i.huppertz等人,methods65,274(2014))。棄去洗滌緩衝液後,將珠與73μl無rnase的水、20μl5×pnk緩衝液ph6.5(350mmtris-hclph6.5,50mmmgcl2,10mmdtt)、5μl10u/μlt4pnk(3'磷酸酶減)(neb)、2μlrnasinplus(promega)混合,並在37℃下孵育20分鐘,每2分鐘在1,200r.p.m.下間歇振蕩5秒。將珠用pnk洗滌緩衝液洗滌一次,並將生物素標記的接頭的5'-末端在100μlpnk反應混合物(73μl無rnase的水,10μl10×pnk緩衝液,10μl10mmatp,5μl10u/μlt4pnk(3'磷酸酶減)(neb),2μlrnasinplus(promega))中在37℃下間歇振蕩1小時進行磷酸化。在磷酸化後,將珠在pnk洗滌緩衝液中洗滌兩次,然後在極度稀釋的條件下在15ml總體積反應物(8.9ml無rnase的水,1.5ml的10mmatp,1.5ml的10×rna連接酶緩衝液,75μl20mg/mlbsa(neb),25μl1mdtt,2.25ml100%dmso,0.75ml10u/μlt4rna連接酶1(neb))中進行鄰位連接以最小化複合體間連接。鄰位連接在37℃下進行1小時,並在16℃下進行過夜,持續旋轉。加入二甲基亞碸(dmso)至15%(v/v)終濃度以刺激高度結構化的rna的連接。
步驟6.期望的rna-rna相互作用的選擇和提取和反轉錄
第二天,通過加入edta至終濃度為25mm並在4℃下旋轉15分鐘來停止連接,以防止當珠集中在管壁上時發生分子間連接。將珠在pbst中洗滌一次。在100μl洗脫緩衝液(100mmtris-hclph7.5,50mmnacl,10mmedta,1%sds,10mmdtt,2.5mmd-生物素(invitrogen))中,通過加熱至95℃5分鐘從鏈黴親和素珠中洗脫兩次蛋白質-rna複合體。將所得溶液合併,與50μl800u/ml蛋白酶(neb)混合,並在55℃下孵育2小時。然後將混合物補充無rnase的水至終體積為400μl。rna在400μl苯酚:氯仿:異戊醇(125:24:1,ph4.5)(ambion)中提取,並在1000r.p.m下振蕩的情況下在37℃下孵育20分鐘。將混合物轉移到2mlmaxtract高密度鎖相凝膠管(qiagen)中,並在室溫下以16,000xg離心5分鐘。通過向相同的maxtract管中加入400μl氯仿並在室溫下以16,000xg離心5分鐘除去殘留的苯酚。離心後,將水相轉移到新管中,通過加入1:9體積的3m乙酸鈉ph5.2、1.5μlglycoblue(ambion)和1ml1:1的乙醇:異丙醇並在-20℃孵育過夜,來析出rna。通過在4℃下以21,000g離心30分鐘使析出的rna沉澱。棄去上清液後,將沉澱物用80%乙醇洗滌兩次,風乾直到乙醇完全蒸發。在此階段純化的rna是沒有接頭的rna(rna1或rna2),與接頭連接但不與其它rna鄰位連接的rna(5'-接頭-rna2),以及5'-rna1-接頭-rna2形式的期望嵌合構建體的混合物。通過選擇生物素標記的接頭可耗盡rna1。因此,非信息性的5'-接頭-rna2被耗盡以及在與t7外切核酸酶的下一個反應中。
6.1.從末端接頭(5'-接頭-rna2)中除去生物素。這是基於t7外切核酸酶的rnaseh活性,其不僅從雙鏈dna除去5'單核苷酸,而且還在rna-dna雜合子的rna鏈上發揮核酸外切活性(k.shinozaki,o.tuneko,nucleicacidsresearch5,4245(1978))。互補的dna寡核苷酸(5'-t*c*g*c*attgcatgggctactagcat(seqidno:2),其中*表示通過t7外切核酸酶阻斷其消化的硫代磷酸酯鍵(t.t.nikiforov,r.b.rendle,m.l.kotewicz,y.h.rogers,genomeresearch3,285(1994)),與rna接頭退火,在rna接頭和互補dna鏈之間產生雙鏈dna-rna雜合子。設計互補dna鏈,以便在退火後,使rna接頭的5'末端嵌入,同時使dna鏈的3'末端突出。然後將退火產物用t7外切核酸酶處理。
將rna沉澱物重懸於17μl無rnase的水、4μl10×nebuffer4,7μl100μm互補dna寡核苷酸中。在70℃下變性5分鐘,然後緩慢地(以-0.1℃/s)將溫度緩降至60℃,在60℃下再孵育5分鐘,然後緩慢冷卻(-0.1℃/s)至37℃,並在37℃下孵育15分鐘,來進行退火。然後將退火的混合物與8μl10u/μlt7外切核酸酶(neb)、4μl1mg/mlbsa混合,並在37℃下孵育30分鐘,在30℃下再孵育30分鐘。使用turbodnase嚴格處理除去dna寡核苷酸以及任何汙染的基因組dna:加入44μl無rnase的水、10μl10×turbodnase緩衝液、6μlturbodnase(invitrogen),所得混合物在37℃下孵育1小時。dnase處理的rna通過如上所述的苯酚:氯仿萃取和乙醇沉澱來純化。
6.2.es-2,mef樣品中通過基於抗體的rna-dna雜合子的耗盡(genereadrrnadepletionkit(qiagen))來除去rrna。根據製造商的說明書伴以下列修改除去rrna。不通過rneasyminelute旋轉柱清除耗盡的rna,這些柱將去除短於200個核苷酸的rna,通過嚴格的dnase處理除去過量的rrna捕獲探針。dnase處理的rna也通過如上所述的苯酚:氯仿提取和乙醇沉澱來純化。
6.3.rna剪切。乙醇沉澱後,通過使用rnaseiii片段化試劑盒根據製造商的方案,將rna片段化為最適用於illuminahiseq的測序的150-400bp的大小範圍。通過2.2×spriselect珠(beckmancoultergenomics)純化片段化的rna並如上所述進行乙醇沉澱。
6.4.與反轉錄適配子連接。接下來,將rna與用作rt反應的引物的3'反轉錄(rt)適配子(/5rapp/agatcggaagagcggttcag/3ddc/(seqidno:3))連接。乙醇沉澱後,將rna沉澱物重懸於20μl的連接反應混合物:1μlrnasinplus(promega),2μl10×rna連接酶緩衝液,7μl20μm預腺苷酸化l3-app適配子,8μl50%peg8000(neb),2μl200u/μlt4rna連接酶2,截短的kq(neb)。將反應物在16℃孵育過夜。
6.5.反轉錄。連接後,通過2×spriselect珠(beckmancoultergenomics)純化rna,並在無rnase的水中洗脫。對於2μg的rna描述以下rt反應,並且對於更高量的rna相應地按比例增加。對於每個實驗或重複,使用含有獨特實驗條形碼序列的不同rt引物。每個rt引物具有5』-/5phos/nnxxxxnnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct(seqidno:4)的形式。根據該方案,每個測序讀段對的第一個讀段包含採用nnnnxxxxnn(seqidno:5)(來自rt引物的反向互補)的構型的條形碼,其中n是用於去除pcr重複的隨機6nt條形碼(g.b.loeb等人,molecularcell48,760(dec14,2012);z.wang等人,plosbiol8,e1000530(2010);j.konig等人,naturestructural&molecularbiology17,909(jul,2010);s.w.chi,j.b.zang,a.mele,r.b.darnell,nature460,479(jul23,2009))。具有相同映射位置和隨機條形碼的任何兩個雙端讀段將僅計數為一個。xxxx是用於多重測序的固定的4nt樣品條形碼(用於es-1的aggt,用於es-2的cgcc,用於es間接的catt,用於mef的cgcc)。任何兩個4nt的樣品條形碼都有不同的三個核苷酸,以避免突變或測序錯誤引起的潛在混淆。
對於cdna合成,將9μlrna與1μl10mmdntp和1μl50μmrt引物混合。將混合物在65℃下加熱5分鐘,並在冰中快速冷卻至少2分鐘。加入4μl5×第一鏈緩衝液(invitrogen),1μldtt0.1m,1μlrnasinplus,1μl10mg/mlt4基因32蛋白(neb)。將所得混合物在50℃下孵育2分鐘,然後加入反轉錄酶,以儘量減少引導錯誤。然後向溶液中加入2μl200u/μlsuperscriptiii反轉錄酶(invitrogen)。然後將rt反應混合物在50℃孵育45分鐘,55℃孵育20分鐘,隨後保持4℃。這裡,為了維持rna-cdna雜合子,省略了反轉錄酶的熱失活。
步驟7.嵌合rna-dna雜合子的生物素下拉
使用鏈黴親和素-生物素親和純化來富集嵌合rna-dna雜合子。這種下拉是在第二次rna片段化和反轉錄之後進行的,以便允許在讀段對的一端大部分測序讀段對覆蓋rna-接頭或接頭-rna接合點。
具體地,50μlmyonec1珠(invitrogen)通過用1×tweenb&w緩衝液(5mmtris-hclph8.0,0.5mmedta,1mnacl,0.05%tween)洗滌兩次,並用1×b&w緩衝液(5mmtris-hclph8.0,0.5mmedta,1mnacl)洗滌一次來製備。然後將珠用100μl2×b&w緩衝液(10mmtris-hclph8.0,1mmedta,2mnacl)重懸。在與100μlc1珠懸浮液合併之前,將rt混合物補充無rnase的水至終體積為100μl,並在rt下孵育30分鐘。將珠回收並用1×b&w緩衝液洗滌三次,然後轉移到新管中,隨後用te緩衝液ph8.0洗滌一次。接下來,通過在37℃下在50μlrnaseh洗脫混合物(39.5μl無rnase的水,5μl10×rnaseh反應緩衝液,0.5μl10%tween-20,5μl5u/μlrnaseh(neb))中完全消化rna鏈1小時,從鏈黴親和素珠釋放cdna鏈。使用磁力收集器在管壁上收集珠,並將上清液收集在新管中用於隨後的操作。通過在70℃加熱20分鐘使rnaseh失活。通過2.2×spriselect珠(beckmancoultergenomics)(v/v)純化cdna。
步驟8.測序文庫的構建
考慮到uv誘導的交聯位點有時會阻止反轉錄,導致缺少5'適配子的截短的cdna(y.sugimoto等人genomebiology13,r67(2012)),採用了允許甚至從截短的cdna構建測序文庫的環化策略(i.huppertz等人,methods65,274(2014))(圖7)。rt引物含有適配子區域以通過illuminapepcr正向引物1.0(5'-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct)(seqidno:6)和pepcr反向引物2.0(5'-caagcagaagacggcatacgagatcggtctcggcattcctgctgaaccgctcttccgatct)(seqidno:7)引發pcr擴增,側翼為bamhi限制性位點和測序條形碼。
8.1.環化。通過circligaseii(epicenter)環化cdna。簡言之,在20μlcircligase反應混合物(12μl無菌水,2μlcircligaseii10×反應緩衝液,1μl50mmmncl2,4μl5m甜菜鹼,1μl100u/μlcircligaseii(epicenter))中將cdna從spriselect珠洗脫,並在60℃下孵育2小時。通過將反應物在80℃下孵育10分鐘使circligaseii失活。
8.2.重線性化。將互補dna寡核苷酸與rt引物退火,產生適合於bamhi限制性位點的短雙鏈區。該策略還阻止bamhi對其他內源性bamhi限制性位點的活性。接下來,應用bamhi,產生在5'末端和3'末端具有適配子的線性cdna,以引發隨後的pcr擴增。接下來,將寡核苷酸退火混合物(43μl水,6μl10×fastdigest緩衝液(fermentas),5μl20μmcut_oligo(5'-gttcaggatccacgacgctcttcaaaa/3invdt/)(seqidno:8)加入到circligaseii反應物中。加熱至95℃2分鐘,然後進行71個循環,每次20秒,從95℃開始並且在每個循環之後將溫度降低1℃,降至25℃並保持在25℃,來進行退火。加入6μlfastdigestbamhi(fermentas),並在37℃孵育30分鐘。通過2×spriselect珠(beckmancoultergenomics)(v/v)純化重線性化的cdna,並在無核酸酶的水中洗脫。
8.3.第一次pcr預擴增和尺寸選擇。首先使用截短型pcr引物(正向引物dp5,5』-cacgacgctcttccgatct(seqidno:9);反向引物dp3,5』-ctgaaccgctcttccgatct)(seqidno:10)),以少量循環(6個循環)進行第一次pcr預擴增單鏈cdna。已經發現,通過在此階段進行尺寸選擇,最終的文庫不容易被不期望的較小尺寸片段(引物二聚體,僅含有條形碼和/或rna接頭的產物)汙染。
使用以下溫度在包含20μlnebnexthigh-fidelity2×pcrmastermix(neb),各0.625μm的dp5/dp3引物的40μl反應物中進行六個循環的pcr:在98℃下初始變性1個循環30秒;6個循環的擴增:98℃下10秒、65℃30秒、72℃30秒;然後在72℃最後延伸5分鐘;並保持在4℃。pcr產物通過1.8×spriselect珠(v/v)純化,並使用e-gelex2%瓊脂糖凝膠(invitrogen)進行尺寸選擇。從凝膠上切下150bp至350bp之間的dna片段,並用minelute凝膠提取試劑盒(qiagen)純化。
8.4.通過雙鏈特異性核酸酶(dsn)方法(h.yi等人,nucleicacidsresearch39,e140(2011))(es-1,es-間接)去除rrna。為了減少來自es-1和es間接文庫的rrnacdna,也使用截短的pcr引物dp5/dp3預擴增ss-cdna。然而,通過1.8×spriselect珠(beckmancoultergenomics)(v/v)純化後,增加pcr循環數直到可獲得80-100ng的cdna。跳過通過瓊脂糖凝膠的尺寸選擇,因為該步驟將大大減少dna的量。將從spriselect珠洗脫的dna與4.5μl雜交緩衝液(2mnacl,200mmhepes,ph8.0)和無菌水(如果需要)混合至終體積為18μl。所得混合物在98℃變性2分鐘,並在熱循環儀上在68℃下再退火5小時。當反應混合物管仍然在熱循環儀中時,將20μl68℃預熱的2×dsn緩衝液(axxora)加入到反應混合物中,通過上下抽吸10次充分混合,並在68℃孵育該反應物10分鐘。加入2μl1u/μldsn酶(axxora),混合,並在68℃孵育25分鐘以上。通過向反應混合物管中加入40μl2×dsn終止液(axxora)終止反應,充分混合併將管轉移到冰上。然後使用1.8×spriselect珠純化反應混合物。
8.5.最終pcr擴增。對使用全長pcr引物pe1.0和2.0(illumina)從先前步驟產生的dna進行pcr擴增。通過用小等分dna運行試驗(pilots)pcr來仔細滴定pcr循環數以避免過度擴增。pcr產物通過1.8×spriselect珠(v/v)純化並尺寸選擇在250-550(120-420bp的嵌入加上~130bp,illuminape1.0/2.0的組合長度)的片段。最終文庫由qubit(invitrogen)和qpcr定量,經生物分析儀(agilenttechnologies)質量檢測,並提交給illuminahiseq平臺上的雙端測序。
rnahi-c中使用的寡核苷酸序列
本方法中使用的定製設計的rna和dna寡核苷酸為:
生物素化的rna接頭(從idt純化的無rnase的hplc):
5'-rcrurarg/ibiodt/rargrcrcrcrarurgrcrararurgrcrgrargrgra-3'(seqidno:11)
含有rna接頭的互補dna鏈(不含rnase的hplc-純化的,來自sigma):
5'-t*c*g*c*attgcatgggctactagcat-3'(seqidno:12)
預先腺苷酸化的rt適配子(無rnase的hplc-純化的,來自idt):
5』-/5rapp/agatcggaagagcggttcag/3ddc/(seqidno:13)
rt引物(由(i.huppertz等人,methods65,274(2014))改編的)(不含rnase的hplc純化的,來自sigma):
用於es-1樣品的rt引物:
5』-/5phos/nnaggtnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct(seqidno:14)
用於es-2和mef樣品的rt引物(在不同通道測序):
5』-/5phos/nncgccnnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct(seqidno:15)
用於es間接樣品的rt引物:
5』-/5phos/nncattnnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct(seqidno:16)
cut_oligo(hplc-純化的,來自idt)
5'-gttcaggatccacgacgctcttcaaaa/3invdt/-3'(seqidno:17)
bamhi限制性位點為下劃線並粗體印刷的部分。
截短的pcr正向引物dp5(hplc-純化的,來自idt):
5』-cacgacgctcttccgatct(seqidno:18)
截短的pcr反向引物dp3(hplc-純化的,來自idt):
5』-ctgaaccgctcttccgatct(seqidno:19)
illuminapepcr正向引物1.0(page-純化的,來自sigma):
5』-aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct(seqidno:20)
illuminapepcr反向引物2.0(page-純化的,來自sigma):
5』-caagcagaagacggcatacgagatcggtctcggcattcc
tgctgaaccgctcttccgatct(seqidno:21)。
計算流程(computationalpipeline)(rna-hic-工具)
rna-hic-工具是用於分析rnahi-c數據的一系列命令行工具。其是用python和r編寫的,由github版本控制。完整的文檔位於http://systemsbio.ucsd.edu/rna-hi-c。流程將雙端測序讀段作為輸入(圖15a)。rna接頭的寡核苷酸序列和用於多重測序的樣品條形碼也提供給流程。主要輸出包括:1.解析的cdna文庫,包括rna1-接頭-rna2形式的嵌合cdna的列表(參見圖7,15c的終產物),2.每種嵌合cdna的rna1和rna2的基因組定位(圖15d),3.從嵌合cdna的統計學富集推斷的相互作用的rna對(圖15e)。分析步驟如下。
1.去除pcr重複
正向讀段(圖15a中的讀段1)在5'端包含4nt樣品條形碼和6nt隨機條形碼。讀段對被分類為另一個讀段對的pcr重複,因此如果兩個讀段對具有相同的序列並且包含相同的條形碼(10nt),則將其丟棄。工具'remove_dup_pe.py'提供此功能,並生成一個包含非重複讀段的fastq/fasta文件,並報告去除的重複數量。
2.將多重測序讀段分配給相應的實驗樣品
工具'split_library_pairend.py'通過將每個讀段中的樣品條形碼與樣品條形碼列表(用戶輸入文本文件)中的樣品條形碼進行匹配,將每個雙端讀段分配給樣品,生成一個分配給每個樣品的讀段的fastq/fasta文件以及未分配讀段的fastq/fasta文件。
3.在測序文庫中回收cdna
該步驟識別每個讀段對的兩端的重疊區域(如果有的話)。它也儘可能地回收測序文庫中cdna的整個序列。
如果存在重疊,則將該讀段對從100bp和200bp之間的cdna(不計算p5和p7的長度)進行測序(類型2,圖32)。在這種情況下,通過將正向讀段(讀段1)與反向讀段(讀段2)的非重疊區域串聯來完整覆蓋cdna的整個序列。
如果cdna短於100bp,則驗證cdna兩端的p5和p7引物的存在(類型1)。棄去不包含p5或p7的cdna(類型4)。
在沒有重疊下,讀段對從長於200bp的cdna進行測序,其序列只能部分回收(類型3,圖32)。
此功能由「recoverfragment.py」實現,其使用本地比對來識別重疊區域。當與讀段長度(每端100bp)相比,重疊小(15bp以下)時,局部比對可能不靈敏。為了克服這種不靈敏性,「recoverfragment.py」在第一次比對之後(align1,圖32)收集讀段對,而無可識別的重疊,將每個讀段截短為其長度的三分之一(在每個讀段的3'處保留33bp),重複局部比對(align4)。
4.解析嵌合cdna
該步驟基於它們的構型對cdna進行分類(圖15c)。這採用完全回收的cdna序列(類型1和類型2,圖32)和部分回收的(類型3)cdna序列,以及接頭序列作為輸入。它識別cdna中接頭的位置,並根據接頭序列的位置產生5類cdna,其中包括:
1.沒有接頭。不包含接頭序列的任何類型1或類型2的cdna均屬於該類別。該類別可以進一步分為三個子集,包括:
a.僅條形碼。整個cdna是10nt條形碼(4nt樣品條形碼+6nt隨機條形碼),最可能是未連接的rt引物汙染的結果。
b.單個rna。整個cdna是rna的連續部分。
c.rna1-rna2。這些可能在接頭連接之前由鄰位連接產生。
包含四個接頭的類別,包括:
2.rna1-接頭-rna2。這些是從期望的嵌合rna產生的。其兩個讀段完全比對兩個不同的rna基因的任意無接頭的類型3的cdna也歸於該類別中。要求rna1和rna2兩側均含有至少5bp序列。
3.接頭-rna2。接頭成功地連接至rna的5'末端,但是鄰位連接不成功。
4.rna1-接頭。接頭連接至rna的3'末端。這可能是由具有3'-oh基團的rna或rna片段產生的,或者在第二次片段化步驟期間從rna1-接頭-rna2嵌合體切掉其它rna(rna2)產生的。
5.僅接頭。整個cdna是條形碼和接頭序列。
該步驟輸出屬於rna1-接頭-rna2類別的cdna的列表。
5.映射到基因組
以下,所有分析基於讀段對的rna1-接頭-rna2類型。首先,丟棄在接頭的rna1或rna2側上含有小於15bp的任何cdna,因為在映射步驟中不可能將15bp以下的序列唯一地映射到基因組。然後使用bowtie版本0.12.7(b.langmead,c.trapnell,m.pop,s.l.salzberg,genomebiology10,(2009))和參數-f-n1-l15-e200-p9–s將接頭每側的兩個rna片段(rna1和rna2)分別映射到小鼠基因組mm9/ncbi37。在「stitch-seq_aligner.py」中執行的該步驟輸出了rna1和rna2唯一地映射到基因組的讀段對。
使用bowtie2(b.langmead,s.l.salzberg,natmethods9,357(apr,2012))的「--靈敏-局部」模式,利用參數「-d15-r2-n0-l20-is,1,0.75」測試潛在的更靈敏的映射方法。這種「多種子比對」使用20bp的種子,允許任何種子中的0個錯配,種子之間9bp間隔多達15次連續的種子延伸嘗試,以及多達2次的「再播種」。事實證明,這種替代策略比bowtie0.12.7鑑定出略少一些的唯一比對。因此,bowtie0.12.7的結果被傳遞到下一步。
6.鑑定相互作用的rna對
注釋從ensembl(釋放67,小鼠ncbim37)檢索,包括mrna,lincrna,rrna,snrna,snorna,mirna,misc_rnas,trna和轉座子的基因。在該分析中,相同轉座子的不同基因組拷貝被認為是不同的基因。從進一步分析中去除映射到rrna的讀段。對每個基因計數唯一比對讀段(來自rna1-接頭-rna2型的rna1或rna2)的數目。將讀段計數小於5的任何基因濾出。接下來,用fisher精確檢驗測試任何兩個基因之間的關聯。無效假設是基因a和基因b獨立地貢獻於測序讀段。另一種假設是它們對讀段計數的貢獻是相關的。ca,cb分別被表示為基因a和基因b的讀段計數,以及i′a,b表示為其中兩個基因共同出現在相同的讀段對上的共同出現的讀段計數。對每個基因對進行fisher精確檢驗,ia,bca,cb,作為檢驗統計,其中是除基因a(基因b)之外的其他基因的讀段計數。對於每個基因對,計算了p值和fdr(benjamini-hochberg程序(y.benjamini,y.hochberg,journaloftheroyalstatisticalsociety.57,289(1995)),該步驟輸出fdr<0.05的基因對和倍數變化(fc)≥3。fc計算為(ia,b+0.5)/(i′a,b+0.5),其中i′a,b為對照樣品(es間接)中共同出現的讀段計數。該步驟在「select_stronginteraction_rna.py」中執行,其輸出強相互作用的rna對,帶有相互作用區域的信息,支持對的數目,有意義的p值,fdr和倍數變化。
7.鑑定rna相互作用位點
rna相互作用位點定義為通常有助於rna-rna相互作用的連續rna區段。rna相互作用位點從rnahi-c數據推斷為具有多重重疊讀段和與其他rna頻繁共同出現(鄰位連接)的連續rna區段。首先,由5個以上的唯一比對讀段覆蓋的任何連續rna區段被鑑定為候選相互作用位點。其次,任何兩個候選位點之間的關聯都用fisher精確檢驗進行了檢驗。無效假設是候選位點a和基因b獨立地貢獻於測序讀段。另一種假設是它們對讀段計數的貢獻是相關的。ca,cb分別表示為候選位點a和b的讀段計數,以及ia,b表示兩個位點共同出現在同一讀段對上的共同出現的讀段計數。對每個位點對進行fisher精確檢驗,ia,b,ca,cb,作為檢驗統計,其中是除a(b)之外的其他候選位點的讀段計數。對於每對候選位點計算p值和fdr(benjamini-hochberg程序)。表現出顯著相關的候選位點(fdr0);
3.靶rna和源rna都出現在agohits-clip(兩個rna的fpkm>0);
4.源rna和靶rna的rnahi-c鑑定的相互作用位點表現出強鹼基配對(p值<0.05,wilcoxon符號秩檢驗,比較每一雙端讀段的rna1和rna2序列之間的結合能與隨機改組的核苷酸序列的結合能)。
總共302個rna-rna相互作用通過了這些過濾。這些相互作用中大多數(79%)的源rna是snorna(表st2)。因此,snorna優先進行功能分析。
假設大量snorna被酶促加工成mirna樣短rna並與mrna相互作用。這一假設得到了919個rnahi-c鑑定的snorna-mrna相互作用的支持,其中mrna和snorna都被ago結合。此外,ago結合的snorna及其相互作用的mrna在es細胞向中內胚層的指導分化期間顯示出反相關表達變化(p.yu等人,spatiotemporalclusteringoftheepigenomerevealsrulesofdynamicgeneregulation.genomeresearch23,352(feb,2013))(圖17b)。另外,與沒有ago結合的相比,ago結合的snorna及其靶mrna表現出更強的鹼基配對(圖17c)。最後,用作參考地,從snorna處理的小rna與mrna的utr區域相互作用。在rna-rna相互作用涉及的497個snorna中,243個與utr區域相互作用,其中在小rna-seq中檢測到223個(92%),表明經歷了酶切(圖17d)。相比之下,與非utr區相互作用的其他254個snorna含有較少的(55%)小rna。此外,相比於與非utr相互作用的snorna,兩倍以上的utro相互作用的sno-sirna是ago結合的(p值<2.2-16,卡方檢驗)。例如,snora14rna靶向mcl1mrna的3'utr(圖19a)。snora14rna(110-135nt)上的相互作用位點與酶處理的小rna(淺紫色道)以及ago結合區(綠色道)精確重疊。snora14rna的酶促處理部分完全位於髮夾環的一側(圖19b),並且對mcl1utr的靶位點顯示出強結合親和力(-60kcal/mol)。經處理的snora14rna的表達與mcl1mrna的表達負相關(圖19c)。總之,該數據表明大量小幹擾rna源自snorna基因,其與es細胞中的900個以上的mrna相互作用。
無擾動的映射體內rna-rna相互作用物組和rna結構
分析整個rna-rna相互作用物組仍然是艱巨的。開發了rnahi-c技術,從而在無任何擾動下在體內映射任何單一蛋白質所包含的rna-rna相互作用。在胚胎幹細胞中系統地映射rna-rna相互作用物組,揭示了46,780個相互作用。使用rap-seq1驗證了7種相互作用。在該相互作用物組中,大多數mirna和lincrna都與一種mrna特異性相互作用,這與目前的「混雜」rna相互作用的教導相矛盾。在長rna之間的相互作用區域觀察到鹼基配對,表明一類以反式作用的調控序列。此外,通過同時揭示單鏈區域的足跡和每個rna的空間鄰位位點,rnahi-c提供了關於rna結構的新信息。該技術大大擴展了rna-rna相互作用物組的可識別部分,而不擾亂rna表達的內源水平。
rnahi-c的模擬分析
數據合成。為了評估rnahi-c的靈敏度和特異性(包括其實驗和計算程序),進行了模擬分析。通過計算模擬數據生成過程,模擬了1,000,000個雙端讀段。用於模擬的參數是從實際數據中得出的。模擬數據生成過程如下。
對於每個雙端讀段(2×100個鹼基):
1.從具有相同概率的四個樣品條形碼選擇樣品條形碼,並將其與6nt隨機條形碼串聯(如圖15a所示)。
2.分別以[0.1、0.3、0.1、0.3、0.2]概率將該雙端讀段分配到[僅接頭、無接頭、rna1-接頭、接頭-rna2、rna1-接頭-rna2]的列表中的一種類型的cdna(如圖15c所示)。
3.如果將此讀段對分配給含接頭的類型,則以相等的概率隨機選擇1或2個接頭。應注意,少量的含接頭的讀段對包含2個接頭;使用相等的概率是估計最壞情況的保守選擇。
4.根據步驟2中確定的cdna類型,產生rna1和rna2部分的序列。對於rna1和rna2兩者,
a.由l~unif(15,150)模擬其長度,
b.基於以下概率從[「mirna」、「mrna、「lincrna」、「snorna」、「snrna」、「trna」]中選擇rna類型:
c如果長度l<50,使用[0.2、0.2、0.1、0.2、0.2、0.1],
d否則,使用[0.05、0.4、0.2、0.2、0.1、0.05];
e.根據ensembl採集的rna型(釋放67,小鼠ncbim37)隨機選擇一種rna,
f.從所選rna隨機取長度l的序列片段。
5.串聯由步驟1、3、4生成的條形碼、接頭和rna片段,產生合成cdna序列。
6.如果步驟5中的合成cdna為100bp以上,則分別從正義鏈和反義鏈的合成cdna的兩端取100個鹼基。
7.如果步驟5中的合成cdna短於100bp,則將其正義鏈和反義鏈分配為正向讀段和反向讀段,並將p5和p7引物序列連接至兩個讀段。
8.在每個鹼基上以0.01的比率模擬測序誤差(n.j.loman等人,performancecomparisonofbenchtophigh-throughputsequencingplatforms.naturebiotechnology30,434(may,2012))。
步驟1-5根據實驗程序模擬了cdna序列,步驟6-8基於該cdna序列模擬了雙端讀段。保持模擬的相互作用的rna對,以及每個部分(rna1、接頭和rna2,如果適用)的cdna類型和長度,用於與計算預測進行比較。
評估中間結果和最終結果。
合成數據用於評估兩個中間分析步驟以及最終預測的靈敏度和特異性。
首先,比較程序鑑定的cdna長度(rna-hic-工具的步驟3的輸出)與實際(合成的)長度(表8)。該步驟「3.在測序文庫中回收cdna」將每個cdna根據它們的長度分配為四種類型,即類型1(200bp);類型4(未知)(圖s32)。該算法實現了對每種類型的鑑定的高靈敏度和特異性。只有很少(0.58%)的短於200bp的cdna被鑑定為超過200bp。這些錯誤是由於正向讀段和反向讀段的小重疊(通常在0到5bps之間),這無法由局部比對所檢測到。
表8.程序鑑定的和真實的cdna長度範圍的比較。將每種類型的程序鑑定的cdna的計數(列1-4)與其真實類型(行)進行比較。
當程序鑑定的長度短於200bp(類型1和類型2)時,可以計算精確長度。在這些情況下,程序鑑定的長度通常與模擬cdna的長度精確匹配(圖33a)。
接下來,比較每個cdna的程序鑑定的嵌合構型(rna-hic-工具的步驟4的輸出)與合成構型。在步驟「4.解析嵌合cdna」中,基於接頭序列的存在,該算法將cdna分為五類。該算法對「rna1-接頭-rna2」形式的cdna靈敏度達到99.89%,特異性為95.82%(表9)。
表9.程序鑑定的和真實的cdna構型的比較。將程序鑑定的構型(列)的cdna計數與其真實構型(行)進行比較。
最後,比較程序預測的和模擬的rna-rna相互作用。模擬數據集包含200,200個嵌合rna對,其中檢測到131,571對rna(靈敏度=65.72%,特異性=92.57%,圖st1-c)。還分別計算了每種類型rna的相互作用的靈敏度和特異性(圖33c)。無論參與的rna類型如何,該方法顯示很少的假陽性(特異性≥90%)。不涉及轉座子rna或snrna的相互作用比那些涉及的顯示出更少的假陰性。這是由於轉座子序列和snrna序列的重複性質。最嚴重的情況涉及linerna,靈敏度下降到52%。據保守估計,涉及轉座子rna的相互作用約有一半可能被該程序錯過。估計約2/3至3/4的不涉及轉座子rna的相互作用將被鑑定出來。
rap-seq驗證
進行小鼠es細胞的malat1rap測序實驗。交聯後,使用5個反義寡核苷酸下拉malat1,然後測序與malat1一起純化的其他rna。進行肌動蛋白rap測序作為對照。malat1rna本身在malat1rap-seq中顯示比肌動蛋白rap-seq增加5.81倍,證實了純化的有效性。rnahi-c報導,malat1作為與tfrc、slc2a3、eif4a2和0610007p14rikrna相互作用的「中樞」lincrna。這些rna在malat1rap-seq中顯示比肌動蛋白rap-seq增加14.6(0610007p14rik)、4.53(slc2a3)、3.38(eif4a2)和2.39(tfrc)倍(最大卡方檢驗p值<0.0003)。這表明來自rnahi-c和malat1rap-seq的malat1靶標的強烈重疊。
對於另一個驗證,進行tfrcrap-seq實驗。tfrc被rnahi-c鑑定為malat1相互作用的rna(圖1d)。問及tfrc下拉是否可以反向鑑別malat1。與肌動蛋白rap-seq相比,tfrcrna本身在tfrcrap-seq中顯示2.87倍增加。在相同的數據集中,malat1rna顯示3.84倍增加,比較tfrcrap-seq與肌動蛋白rap-seq(p值<2.2×10-16,來自檢驗無效假設倍數變化=1)。
檢查與由rnahi-c鑑定的tfrc相互作用的其它rna,並且也可以由tfrcrap-seq驗證。rnahi-c數據鑑定了與tfrc相互作用的總共5種rna。除了malat1,其餘四個都是snorna,即snord13,snora3,snord52,snora74。與肌動蛋白rap-seq相比,這4種snorna中的3種在tfrcrna-seq中顯示出倍數增加(snord13為1.4倍,snora3為13.6倍,snora74為8.7倍),證實了這些相互作用(卡方檢驗p值<0.00002)。總之,rap-seq證實了幾乎所有的rnahi-c鑑定的相互作用。通過兩種類型的實驗(rnahi-c和rap-seq),幾種rna相互作用(如上所述)在小鼠es細胞中被記為「真實的」。
snorna-mrna相互作用與mrna假尿苷的比較
將假尿苷酸化測序數據(ψ-seq)與rna-相互作用位點進行比較。schwartz等人在酵母和小鼠骨髓來源的樹突狀細胞(bmddc)中進行ψ-seq。檢索bmddcψ-seq數據(cmc處理的gsm1464234和對照gsm1464235),並使用文中描述的生物信息學過程稱為假尿苷(ψ位點)。簡言之,ψ位點被確定為在正確的鏈和方向上具有超過5個經cmc處理的讀段位於'u'旁邊並且具有大於3的ψ-fc值。這產生了在總數8,194,131個'u'位中的386個ψ-位點(0.00471%u是ψ位點)。
接下來,比較這386個位點與rnahi-c鑑定的rna相互作用位點。已經認識到ψ-seq和rnahi-c在不同的細胞類型中進行。然而,在rna相互作用的位點中,共551,634個u中有93個是ψ位點(0.0109%)。因此,通過rnahi-c確定的rna相互作用位點富集有ψ位點(優勢率=4.4,卡方檢驗p值=7.70×10-95)。
此外,詢問是否在由rnahi-c檢測的snorna-mrna相互作用位點中富集了ψ位點。在snorna參與的相互作用位點中,總共136,535個u(0.0381%)中共有57個ψ位點。與整個轉錄組相比,rnahi-c檢測到的snorna參與的相互作用位點大量富集有ψ位點(優勢率=10.2,卡方檢驗p值<1×10-100)。雖然已知snorna會促進rna假尿苷化,但這些數據表明哪些snorna可能是專門負責的。(表10)。
表10ψ位點和rna相互作用位點關聯檢驗的雙向相依表。
rna分子之間的相互作用發揮關鍵的調控作用,並且通常由rna結合蛋白介導(ray,d.等人acompendiumofrna-bindingmotifsfordecodinggeneregulation.nature499,172-177,doi:10.1038/nature12311(2013)),如argonaute蛋白(ago)、pum2、qki和snornp蛋白(meister,g.argonauteproteins:functionalinsightsandemergingroles.natrevgenet14,447-459,doi:10.1038/nrg3462(2013);hafner,m.等人transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010);granneman,s.,kudla,g.,petfalski,e.&tollervey,d.identificationofproteinbindingsitesonu3snornaandpre-rrnabyuvcross-linkingandhigh-throughputanalysisofcdnas.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica106,9613-9618,doi:10.1073/pnas.0901997106(2009))。儘管最近有進展,例如par-clip4,hits-clip6和clash7、8,但映射所有蛋白質輔助的rna-rna相互作用仍然是艱巨挑戰(hafner,m.等人transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010);chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009);helwak,a.,kudla,g.,dudnakova,t.&tollervey,d.mappingthehumanmirnainteractomebyclashrevealsfrequentnoncanonicalbinding.cell153,654-665,doi:10.1016/j.cell.2013.03.043(2013).kudla,g.,granneman,s.,hahn,d.,beggs,j.d.&tollervey,d.cross-linking,ligation,andsequencingofhybridsrevealsrna-rnainteractionsinyeast.procnatlacadsciusa108,10010-10015,doi:10.1073/pnas.1017386108(2011))。在這三種方法的每一種中,每個實驗只能分析由一種rna結合蛋白介導的相互作用。另外,每個實驗需要蛋白質特異性抗體(hits-clip或par-clip)或轉化細胞系中標記蛋白質的穩定表達(clash)。
早期方法通常需要所提出的相互作用的一個或幾個組分的異位表達。這些方法包括螢光素酶報告分析和使用合成rna模擬物進行靶捕獲(nicolas,f.e.experimentalvalidationofmicrornatargetsusingaluciferasereportersystem.methodsinmolecularbiology732,139-152,doi:10.1007/978-1-61779-083-6_11(2011);lal,a.等人,captureofmicrorna-boundmrnasidentifiesthetumorsuppressormir-34aasaregulatorofgrowthfactorsignaling.plosgenet7,e1002363,doi:10.1371/journal.pgen.1002363(2011))。因為異位表達很少再現內源性表達水平,因此謹慎地將這些方法的結果解釋為潛在的相互作用而不是體內相互作用。要注意的是,mirna傾向於「混雜地」與許多mrna相互作用的前提主要來源於使用異位表達的數據(du,t.&zamore,p.d.beginningtounderstandmicrornafunction.cellres17,661-663,doi:10.1038/cr.2007.67(2007)).。
開發rnahi-c方法檢測體內蛋白質輔助的rna-rna相互作用。在該程序中,rna分子與其結合的蛋白質交聯,然後連接至生物素化的rna接頭,使得由相同蛋白質共同結合的rna分子形成rna1-接頭-rna2形式的嵌合rna。使用鏈黴親和素包被的磁珠分離這些含接頭的嵌合rna,並進行雙端測序(方法,圖1a,圖7)。因此,每個非冗餘雙端讀段反映了分子相互作用。該技術的一些設計方面受到染色體構象捕獲方法的啟發(kalhor,r.,tjong,h.,jayathilaka,n.,alber,f.&chen,l.genomearchitecturesrevealedbytetheredchromosomeconformationcaptureandpopulation-basedmodeling.naturebiotechnology30,90-98,doi:10.1038/nbt.2057(2012);belton,j.m.等人,hi-c:acomprehensivetechniquetocapturetheconformationofgenomes.methods58,268-276,doi:10.1016/j.ymeth.2012.05.001(2012))。
rnahi-c方法提供了映射rna-rna相互作用的數個優點。首先,rnahi-c直接分析內源細胞特徵,而不在交聯前引入任何外源核苷酸或蛋白質編碼基因(hafner,m.等人,transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010);helwak,a.,kudla,g.,dudnakova,t.&tollervey,d.mappingthehumanmirnainteractomebyclashrevealsfrequentnoncanonicalbinding.cell153,654-665,doi:10.1016/j.cell.2013.03.043(2013);lal,a.等人captureofmicrorna-boundmrnasidentifiesthetumorsuppressormir-34aasaregulatorofgrowthfactorsignaling.plosgenet7,e1002363,doi:10.1371/journal.pgen.1002363(2011);baigude,h.,ahsanullah,li,z.,zhou,y.&rana,t.m.mir-trap:abenchtopchemicalbiologystrategytoidentifymicrornatargets.angewchemintedengl51,5880-5883,doi:10.1002/anie.201201512(2012))。這消除了通過改變rna或蛋白質表達水平產生的報告偽相互作用的不確定性。此外,它使rnahi-c非常適合於測定組織樣品。第二,使用生物素化的接頭作為選擇標記可以避免對於蛋白質特異性抗體的需求或表達標記蛋白的需要。這允許rna-rna相互作用物組的無偏倚映射。如文獻中所述,其它方法一次只能對一種rna結合蛋白起作用。第三,僅捕獲由相同的單個蛋白質分子聚集的rna,避免捕獲單獨結合至相同蛋白質的不同拷貝的獨立的rna分子(可能導致報告偽相互作用)(hafner,m.等人,transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010);chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009))。第四,通過在極度稀釋的條件下在鏈黴親和素珠上進行rna連接步驟,使得通過rna隨機連接至其它附近rna產生的假陽性最小化。第五,rna接頭提供了一個清晰的邊界,描繪跨越連接位點的測序讀段,從而避免了映射測序讀段的含糊不清。第六,通過在pcr擴增前將隨機6個核苷酸條形碼連接至每個嵌合rna中,隨後僅一次計數與相同的條形碼完全重疊的測序讀段,來去除潛在的pcr擴增偏倚(chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009),loeb,g.b.等人,transcriptome-widemir-155bindingmaprevealswidespreadnoncanonicalmicrornatargeting.molcell48,760-770,doi:10.1016/j.molcel.2012.10.002(2012);wang,z.等人,iclippredictsthedualsplicingeffectsoftia-rnainteractions.plosbiol8,e1000530,doi:10.1371/journal.pbio.1000530(2010);konig,j.等人,icliprevealsthefunctionofhnrnpparticlesinsplicingatindividualnucleotideresolution.natstructmolbiol17,909-915,doi:10.1038/nsmb.1838(2010))。
在具有較小技術差異的小鼠胚胎幹(es)細胞(其被稱為es-1和es-2)上進行了兩個獨立的rnahi-c測定(表5,圖9-12)。使用兩種交聯劑(甲醛和egs),其「有效捕獲通過多重蛋白質中間體間接連接的rna」1(es間接),產生用於間接rna相互作用的文庫((engreitz,j.m.等人,rna-rnainteractionsenablespecifictargetingofnoncodingrnastonascentpre-mrnasandchromatinsites.cell159,188-199,doi:10.1016/j.cell.2014.08.018(2014);nowak,d.e.,tian,b.&brasier,a.r.two-stepcross-linkingmethodforidentificationofnf-kappabgenenetworkbychromatinimmunoprecipitation.biotechniques39,715-725(2005);zeng,p.y.,vakoc,c.r.,chen,z.c.,blobel,g.a.&berger,s.l.invivodualcross-linkingforidentificationofindirectdna-associatedproteinsbychromatinimmunoprecipitation.biotechniques41,694-698(2006);zhao,j.等人,genome-wideidentificationofpolycomb-associatedrnasbyrip-seq.molcell40,939-953,doi:10.1016/j.molcel.2010.12.011(2010))。從小鼠胚胎成纖維細胞(mef)和小鼠腦中產生另外兩個獨特的文庫,為生物信息學質量評估提供兩個額外的數據集(圖13)。證實每個文庫含有期望形式(rna1-接頭-rna2)和長度的rna構建體(圖1b)。對每個文庫測序,平均得到4730萬個雙端讀段,其中大約1510萬個非冗餘雙端讀段代表期望的嵌合形式(圖1c)。此外,進行了三個對照實驗。第一和第二對照實驗分別排除了交聯步驟(非交聯對照)和蛋白質生物素化步驟(非生物素化對照)(rnahi-c的對照實驗)。第三個對照實驗使用果蠅s2細胞和小鼠es細胞來測試rna隨機連接的程度(跨物種對照)。交聯後,來自兩個細胞系的裂解物在蛋白質生物素化和鄰位連接之前混合。將混合物進行實驗程序的其餘部分,並產生測序文庫(fly-mm)。映射到兩個物種(假陽性)的rna對的比例為0.52%。然而,當es-1測序文庫進行相同的信息學分析時,將0.55%rna對映射到兩個物種(小鼠和果蠅基因組),表明實驗性假陽性(可能是由於隨機連接)與信息學程序的誤差範圍相比較不頻繁(rnahi-c的對照實驗)。
表5:rnahi-c樣品的描述。「讀段對的總數」是每個樣品的雙端測序讀段的數量。「rna1-接頭-rna2」形式的非重複讀段對的數量是生物信息學流程的步驟4(其解析嵌合cdna)的輸出中的雙端讀段的數量。
創建了一套生物信息學工具(rna-hic-工具)來分析和顯現rnahi-c數據(圖14-15)。rna-hic-工具自動化分析步驟,包括去除pcr重複,分離復用樣品,鑑定接頭序列,分離連接點讀段,召集相互作用的rna,進行統計學評估,分類rna相互作用類型,召集相互作用位點和分析rna結構(方法)。它還為rna-rna相互作用物組和rna內的鄰位位點提供可視化工具(圖16)。
比較5種rnahi-c文庫。es-1和es-2最相似,其由fpkms的相關性判斷(對於接頭的左側和右側的讀段片段分別計算的),接著是es-間接,然後是mef和腦組織(圖13)。從es-1和es-2鑑定的相互作用的rna對表現出強重疊(p值<10-35,置換檢驗)(表6)。在mef中鑑定的相互作用與es樣品中鑑定的沒有顯著的重疊(每個重疊的p值=1,置換檢驗)。例如,trim25rna的3'utr與小核仁rna(snorna)snora1之間的相互作用分別由es-1和es-2樣品中的24個和22個雙端讀段支持,但在es-間接中(雙重交聯和uv交聯之間的差異)或mef文庫未檢測到(圖1c)。包括snora1的多達172個snorna被鑑定為與agohits-clip數據(綠色泳道,圖1c)和酶促處理的小rna(紅色泳道,圖1c,圖17-19)中檢測到的mrna相互作用(yu,p.等人,spatiotemporalclusteringoftheepigenomerevealsrulesofdynamicgeneregulation.genomeres23,352-364,doi:10.1101/gr.144949.112(2013).)。這支持了snorna基因的轉錄物可以酶促加工成mirna樣小rna並與risc複合體中的mrna相互作用的提議(ender,c.等人,ahumansnornawithmicrorna-likefunctions.molcell32,519-528,doi:10.1016/j.molcel.2008.10.017(2008);brameier,m.,herwig,a.,reinhardt,r.,walter,l.&gruber,j.humanboxc/dsnornaswithmirnalikefunctions:expandingtherangeofregulatoryrnas.nucleicacidsres39,675-686,doi:10.1093/nar/gkq776(2011))。(具有mirna樣相互作用的其它rna)。
表6.映射到兩個基因組的讀段對的分布。不包括在本表中的讀段不能映射到任何基因組或將相同的rna部分映射到兩個基因組。rna部分是接頭序列任一側的讀段序列。
合併es-1和es-2文庫以推斷es細胞中的rna-rna相互作用物組。這些數據包括454萬個非重複的雙端讀段,將其明確地分成兩個rna片段,兩個片段都獨特地映射到基因組(mm9)。鑑定了46,780個rna間相互作用(fdr<0.05,fisher精確檢驗和benjamin&hochberg校正)(圖20)。如預期的,rna表達水平(fpkm)與每個rna上的rnahi-c讀段的數量微弱相關,但fpkm與相互作用的統計學顯著性(fdr)無關(圖20c-d)。mrna-snorna相互作用是最豐富的類型,儘管還檢測到數千個mrna-mrna和數百個lincrna-mrna,假基因rna-mrna,mirna-mrna相互作用(圖21)。這可能是任何生物體中描述的第一個rna-rna相互作用物組。對於整個實驗和分析程序我們的模擬表明了大約66%的靈敏度和93%的特異性(rnahi-c的模擬分析)。
為了確認更大規模的相互作用,進行rna反義寡核苷酸純化測序(rap-seq)(engreitz,j.m.等人,rna-rnainteractionsenablespecifictargetingofnoncodingrnastonascentpre-mrnasandchromatinsites.cell159,188-199,doi:10.1016/j.cell.2014.08.018(2014))。首先,進行malat1rap-seq和actbrap-seq(對照)以檢測涉及malat1的相互作用(snorna-mrna相互作用與mrna假尿苷的比較)。malat1rna本身在malat1rap-seq中顯示出比actarap-seq增加5.81倍,證實了純化的有效性。rna-hic報告的malat1相互作用的rna(圖1d)顯示,在malat1rap-seq中比actarap-seq增加14.6(0610007p14rik)、4.53(slc2a3)、3.38(eif4a2)和2.39(tfrc)倍(p值<0.0003,卡方檢驗)。這表明malat1靶標在rnahi-c和malat1rap-seq中的強烈重疊。接下來,問及tfrcrap是否通過tfrcrap-seq反向鑑定malat1(snorna-mrna相互作用與mrna假尿苷的比較)。tfrcrna本身在tfrcrap-seq中顯示相比於actbrap-seq的2.87倍增加。malat1表現出3.84倍的增加(p值<2.2×10-16,來源於檢驗無效假設倍數變化=1)。此外,由rnahi-c鑑定的四種其他tfrc相互作用的rna中,有三種顯示出1.4-13.6倍的增加(p值<0.00002,卡方檢驗)。總之,rap-seq驗證了另外7種rnahi-c鑑定的相互作用。
已經報導了rna-rna相互作用是「令人驚訝的混雜」(du,t.&zamore,p.d.beginningtounderstandmicrornafunction.cellres17,661-663,doi:10.1038/cr.2007.67(2007))。提示一種細胞類型中每個mirna與300至1,000個mrna相互作用,對lincrna也提出了類似的內容(chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009);guttman,m.等人,chromatinsignaturerevealsoverathousandhighlyconservedlargenon-codingrnasinmammals.nature458,223-227,doi:10.1038/nature07672(2009))。然而,觀察到的rna-rna相互作用物組(46,780個相互作用)是無標度的網絡,其程度分布符合冪定律(圖1d,圖34)(barabasi,a.l.&oltvai,z.n.networkbiology:understandingthecell'sfunctionalorganization.natrevgenet5,101-113,doi:10.1038/nrg1272(2004))。換句話說,參與rna-rna相互作用的大多數rna具有特定的相互作用伴侶,並且具有給定數量的相互作用伴侶的rna的數量隨著相互作用伴侶數量的增加而呈指數級降低。如果相互作用僅限於mrna,lincrnas,mirnas,假基因rna和反義轉錄物,則這種全局性質不會改變(圖1d)。此外,源自小鼠腦的rna-rna相互作用物組(57,833個相互作用)是無標度的(圖34b),表明這種全局性質不是細胞類型特異的。在每種細胞類型中,絕大多數mirna和lincrna與1至3個mrna相互作用,其中超過80%與一種mrna特異性相互作用(圖1e)。總而言之,「混雜」的rna是來源於rnahi-c的rna-rna相互作用物組中的特例。據推測,這是因為與以前的方法不同,rnahi-c直接捕獲在內源性細胞條件下與每個單獨的蛋白質分子共連接的rna分子。
大部分(83.05%)的相互作用的rna顯示重疊的rnahi-c讀段(圖3a),表明相互作用通常集中在rna的特定區段。鑑定重疊讀段片段的「峰」,稱為「相互作用位點」(圖3b)。相互作用位點不僅出現在mirna(整個成熟mirna)、mrna、lincrna上,而且也出現在假基因和轉座子rna上(圖3c)。在l1、sine、ervk、malr和erv1轉座子rna中存在超過2000個相互作用位點(表7),表明它們與其他rnas頻繁相互作用(shalgi,r.,pilpel,y.&oren,m.repressionoftransposable-elements-amicrornaanti-cancerdefensemechanism?trendsingenetics:tig26,253-259,doi:10.1016/j.tig.2010.03.006(2010);yuan,z.,sun,x.,liu,h.&xie,j.micrornagenesderivedfromrepetitiveelementsandexpandedbysegmentalduplicationeventsinmammaliangenomes.plosone6,e17666,doi:10.1371/journal.pone.0017666(2011)。另外,假尿苷在snorna-mrna相互作用的mrna相互作用位點富集,證實了某些rna區段在某些類型的rna相互作用中是有利的(schwartz,s.etal.transcriptome-widemappingrevealswidespreaddynamic-regulatedpseudouridylationofncrnaandmrna.cell159,148-162,doi:10.1016/j.cell.2014.08.028(2014))。
表7.不同類型基因和轉座子中相互作用位點的分布。新:未注釋的基因組區域。
問及鹼基互補是否被不同類型的rna-rna相互作用所利用。通過連接片段對(rna1,rna2)的平均雜交能估計一對相互作用的rna的雜交能,並將其與由鹼基的隨機改組產生的對照rna的雜交能進行比較(ray,d.等人,acompendiumofrna-bindingmotifsfordecodinggeneregulation.nature499,172-177,doi:10.1038/nature12311(2013);bellaousov,s.,reuter,j.s.,seetin,m.g.&mathews,d.h.rnastructure:webserversforrnasecondarystructurepredictionandanalysis.nucleicacidsresearch41,w471-w474,doi:doi10.1093/nar/gkt290(2013))。互補鹼基在幾乎所有類型的rna-rna相互作用中都是優選的,並且在轉座子rna-mrna,mrna-mrna,假基因rna-mrna,lincrna-mrna,mirna-mrna相互作用中最顯著(p值<2.4-18),但是在ltr-假基因rna相互作用中沒有觀察到(圖3d,圖24)。這個數據表明一種新的機制,其中鹼基配對有助於長rna中序列特異性的轉錄後調控。
如果這些rna-rna相互作用是序列特異性的,則rna相互作用位點應該處於選擇壓力下(gong,c.&maquat,l.e.lncrnastransactivatestau1-mediatedmrnadecaybyduplexingwith3'utrsviaaluelements.nature470,284-288,doi:10.1038/nature09701(2011))。已經發現,在相互作用位點處,種間保守水平強烈增加,保守峰精確地確定了兩個rna片段的連接點(圖3d)(cooper,g.m.等人,distributionandintensityofconstraintinmammaliangenomicsequence.genomeres15,901-913,doi:10.1101/gr.3577405(2005))。當與lincrna,假基因rna,轉座子rna或其他mrnas相互作用時,mrna上的相互作用位點比其餘轉錄物更保守(圖25)。lincrna和假基因rna的相互作用位點在lincrnas-mrna,假基因rna-mrna和假基因rna-轉座子rna相互作用中表現出增加的保守性(圖25)。相互作用位點上增加的保守性不是由於外顯子-內含子的邊界(圖26)。總之,鹼基互補在長rna的相互作用中廣泛傳播。互補區域在進化上是保守的。
雖然設計的rnahi-c最初用於映射分子間相互作用,但是發現rnahi-c顯示rna二級結構和三級結構。以上所有分析均基於分子間讀段。通過觀察分子內讀段,了解rna結構的兩個特徵。首先,通過rnasei消化位點的密度(連接前應用rnasei消化,參見圖1a中的步驟2,圖27)鑑定rna的單鏈區域的足跡。第二,通過鄰位連接捕獲每個rna的空間鄰位位點(圖1a中的步驟5)。總共67,221個讀段對映射到單個基因,但沒有映射到彼此或相同的鏈上的2,000bp內,因此是從分子內切割和連接產生的(圖28)。通過將測序讀段中的rna1和rna2的取向與其在基因組中的取向相比較,每個切割和連接的序列可以明確地分配給兩個結構類之一(圖4a)。這些讀段提供2,374個rna的空間鄰近信息,包括來自1,696個已知基因和678個新基因的rna。例如,從snora73轉錄物產生277個切割和連接的序列(圖4b)。rnasei消化位點的密度(圖4c)強烈地預測了rna的單鏈區域(熱圖,圖4e)。檢測到6對鄰位位點(圓形,圖4d)。每一對由三個以上的具有重疊連接位置的切割和連接的序列(黑點,圖4b)支持。六個鄰位位點對中的五個在通常接受的二級結構中物理上接近(相同顏色的箭頭,圖4e)。在snora14上,根據測序推斷的二級結構,一對推斷的鄰位位點出現較遠(圖29)。然而,核糖核蛋白dyskerin在體內彎曲snora14轉錄物,使得兩個假尿苷酸化環彼此接近,如通過切割和連接的序列所預測的(箭頭,圖4f)(kiss,t.,fayet-lebaron,e.&jady,b.e.boxh/acasmallribonucleoproteins.molcell37,597-606,doi:10.1016/j.molcel.2010.01.032(2010))。結構信息甚至可以在新的轉錄物和mrna的一些部分得到(圖30,31)。迄今為止,解析任何單個rna的空間鄰位鹼基仍然是一項艱巨的挑戰。es細胞中的rnahi-c提供數千個rna的分子內空間鄰近信息。此外,每個rna的單鏈足跡同時映射。因此,rnahi-c大大擴大了我們檢測rna結構的能力。
映射rna相互作用的關鍵是選擇。在rnahi-c中引入可選擇的接頭使得能夠無偏倚地選擇相互作用的rna,使得可以全局地映射rna相互作用物組。es細胞中每個rna的相互作用伴侶的數量是非常不平衡的,導致無標度的rna網絡。長rna之間的相互作用經常使用一小部分轉錄物。類似於蛋白質相互作用域,提出了rna相互作用位點的概念。rna相互作用位點利用鹼基配對來促進長rna的相互作用,提示了一種新型的反式調控序列。這些反式調控序列比轉錄物的其他部分更進化保守。rna結構也可以通過rnahi-c進行映射。本文提供了其中rna被蛋白質彎曲的示例性實施方案,並且通過rnahi-c的分子內讀段顯示這種三級結構。因此,這種方法和數據應該大大有助於將來對rna功能和調控作用的調查。
軟體訪問
rna-hic-工具軟體可以在http://systemsbio.ucsd.edu/rna-hi-c獲得。
從上述可以理解,為了說明的目的,本文已經描述了本公開的各種實施方案,並且在不脫離本公開的範圍和精神的情況下可以進行各種修改。因此,本文公開的各種實施方案並不意圖是限制性的,其真實範圍和精神由所附權利要求表示。
附加實施例
在一些實施方案中,一種用於產生包含在細胞中彼此相互作用的rna的嵌合rna的方法,其中所述方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,rna與蛋白質的所述交聯在完整細胞上或在細胞裂解物中進行。在一些實施方案中,所述交聯包括uv交聯。在一些實施方案中,所述方法還包括將所述蛋白質與有助於所述蛋白質在表面上固定化的試劑相關聯。在一些實施方案中,促進固定化的所述試劑包括生物素。在一些實施方案中,蛋白質至少一個半胱氨酸被生物素化。在一些實施方案中,該方法還包括將與相同蛋白質分子交聯的所述rna片段化。在一些實施方案中,所述片段化包括在有助於所述rna部分消化的條件下將與相同蛋白質分子交聯的所述rna與rnase接觸。在一些實施方案中,所述方法還包括將與相同蛋白質分子交聯的所述rna連接至有助於所述rna的回收的試劑。在一些實施方案中,所述連接包括將所述rna的末端連接至所述試劑。在一些實施方案中,將rna與生物素標記的rna接頭連接。在一些實施方案中,生物素標記的rna接頭是2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18.19,20,21,22,23,24,25,26,27,28,29或30個核苷酸長度或任何上述值之間的任何長度。在一些實施方案中,有助於所述rna回收的所述試劑包含核酸。在一些實施方案中,所述核酸包括其上具有生物素的核酸。在一些實施方案中,所述其上具有生物素的核酸與所述rna的所述末端的所述連接包括在將與相同蛋白質分子交聯的所述rna連接在一起以形成嵌合rna之前將其上具有生物素的所述核酸連接至所述rna的5'末端。在一些實施方案中,所述方法還包括從所述嵌合rna的5'區域除去所述生物素。在一些實施方案中,所述方法還包括回收所述嵌合rna。在一些實施方案中,所述方法還包括將所述嵌合rna片段化。在一些實施方案中,該方法還包括dnase處理以消除dna汙染。在一些實施方案中,所述嵌合rna的所述片段化包括在有助於所述rna部分消化的條件下使所述嵌合rna與rnase接觸。在一些實施方案中,所述方法還包括反轉錄所述嵌合rna以產生嵌合cdna。在一些實施方案中,所述方法還包括測定所述嵌合rna或嵌合cdna中源自所述嵌合rna或嵌合cdna中的每個rna的至少一部分序列。在一些實施方案中,該方法還包括鑑定存在於所述嵌合rna中的rna,從而鑑定在細胞中彼此相互作用的rna。在一些實施方案中,鑑定出細胞中至少100個、至少500個、至少1000個或多於1000個rna-rna相互作用。在一些實施方案中,鑑定出在細胞中彼此相互作用的基本上所有的rna。在一些實施方案中,鑑定出細胞中至少70%、至少80%、至少90%或超過90%的直接rna-rna相互作用。在一些實施方案中,在細胞中彼此相互作用的rna的鑑定包括使用自動測序裝置對所述嵌合rna進行序列讀段。在一些實施方案中,在細胞中彼此相互作用的rna的所述鑑定包括從所有序列讀段鑑定嵌合序列。在一些實施方案中,所述方法還包括使用計算機將嵌合rna轉化為注釋的rna簇。在一些實施方案中,所述方法還包括使用由計算機執行的統計檢驗來鑑定所述rna簇之間的直接相互作用。
在一些實施方案中,提供了分離的複合體。分離的複合體可以包含與蛋白質交聯的嵌合rna,其中所述嵌合rna包含在細胞中彼此相互作用的rna。分離的複合體還可以包括包含蛋白質和核酸的複合體,中間體蛋白質和核酸,或蛋白質複合體和核酸,其中所述核酸是rna。在一些實施方案中,分離的複合體包括包含蛋白質和核酸的複合體,中間體蛋白質和核酸,或蛋白質複合體和核酸,其中所述核酸是rna。
在一些實施方案中,提供了用於鑑定候選治療劑的方法,其中所述方法包括使用本文所述任何實施方案的方法鑑定細胞中彼此相互作用的rna,並評估試劑減少或增加所述rna的相互作用的能力,其中如果所述試劑能夠減少或增加所述rna的相互作用,則所述試劑是候選治療劑。在一些實施方案中,用於鑑定在細胞中彼此相互作用的rna的方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,所述rna與蛋白質的交聯在完整細胞上或在細胞裂解物中進行。在一些實施方案中,所述交聯包括uv交聯。在一些實施方案中,所述方法還包括將所述蛋白質與有助於所述蛋白質在表面上固定化的試劑相關聯。在一些實施方案中,有助於固定化的所述試劑包括生物素。在一些實施方案中,該方法還包括將與相同蛋白質分子交聯的所述rna片段化。在一些實施方案中,所述片段化包括在有助於所述rna部分消化的條件下將與相同蛋白質分子交聯的所述rna與rnase接觸。在一些實施方案中,所述方法還包括將與相同蛋白質分子交聯的所述rna連接至有助於所述rna的回收的試劑。在一些實施方案中,所述連接包括將所述rna的末端連接至所述試劑。在一些實施方案中,有助於所述rna回收的所述試劑包括核酸。在一些實施方案中,所述核酸包括其上具有生物素的核酸。在一些實施方案中,所述其上具有生物素的核酸與所述rna的所述末端的所述連接包括在將與相同蛋白質分子交聯的所述rna連接在一起以形成嵌合rna之前將其上具有生物素的所述核酸連接至所述rna的5'末端。在一些實施方案中,所述方法還包括從所述嵌合rna的5'區域除去所述生物素。在一些實施方案中,所述方法還包括回收所述嵌合rna。在一些實施方案中,所述方法還包括將所述嵌合rna片段化。在一些實施方案中,所述嵌合rna的所述片段化包括在有助於所述rna部分消化的條件下使所述嵌合rna與rnase接觸。在一些實施方案中,所述方法還包括反轉錄所述嵌合rna以產生嵌合cdna。在一些實施方案中,所述方法還包括測定所述嵌合rna或嵌合cdna中源自所述嵌合rna或嵌合cdna中的每個rna的至少一部分序列。在一些實施方案中,該方法還包括鑑定存在於所述嵌合rna中的rna,從而鑑定在細胞中彼此相互作用的rna。在一些實施方案中,鑑定出細胞中至少100個、至少500個、至少1000個或多於1000個rna-rna相互作用。在一些實施方案中,鑑定出在細胞中彼此相互作用的基本上所有的rna。在一些實施方案中,鑑定出細胞中至少70%、至少80%、至少90%或超過90%的直接rna-rna相互作用。在一些實施方案中,在細胞中彼此相互作用的rna的鑑定包括使用自動測序裝置對所述嵌合rna進行序列讀段。在一些實施方案中,在細胞中彼此相互作用的rna的所述鑑定包括從所有序列讀段鑑定嵌合序列。在一些實施方案中,所述方法還包括使用計算機將嵌合rna轉化為注釋的rna簇。在一些實施方案中,所述方法還包括使用由計算機執行的統計檢驗來鑑定所述rna簇之間的直接相互作用。在一些實施方案中,所述試劑包括核酸。在一些實施方案中,所述試劑包括化學化合物。
在一些實施方案中,提供了製備藥物的方法,其中所述方法包括將使用本文所述任何實施方案的方法鑑定的試劑配製在藥學上可接受的載體中。在一些實施方案中,通過用於鑑定候選治療劑的方法來配製所鑑定的試劑,其中所述方法包括使用本文所述的任何實施方案的方法鑑定在細胞中彼此相互作用的rna,並評估其用於減少或增加所述rna的相互作用的能力,其中如果所述試劑能夠減少或增加所述rna的相互作用,則所述試劑是候選治療劑。在一些實施方案中,用於鑑定在細胞中彼此相互作用的rna的方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,所述rna與蛋白質的交聯在完整細胞上或在細胞裂解物中進行。在一些實施方案中,所述交聯包括uv交聯。在一些實施方案中,所述方法還包括將所述蛋白質與有助於所述蛋白質在表面上固定化的試劑相關聯。在一些實施方案中,有助於固定化的所述試劑包括生物素。在一些實施方案中,該方法還包括將與相同蛋白質分子交聯的所述rna片段化。在一些實施方案中,所述片段化包括在有助於所述rna部分消化的條件下將與相同蛋白質分子交聯的所述rna與rnase接觸。在一些實施方案中,所述方法還包括將與相同蛋白質分子交聯的所述rna連接至有助於所述rna的回收的試劑。在一些實施方案中,所述連接包括將所述rna的末端連接至所述試劑。在一些實施方案中,有助於所述rna回收的所述試劑包括核酸。在一些實施方案中,所述核酸包括其上具有生物素的核酸。在一些實施方案中,所述其上具有生物素的核酸與所述rna的所述末端的所述連接包括在將與相同蛋白質分子交聯的所述rna連接在一起以形成嵌合rna之前將其上具有生物素的所述核酸連接至所述rna的5'末端。在一些實施方案中,所述方法還包括從所述嵌合rna的5'區域除去所述生物素。在一些實施方案中,所述方法還包括回收所述嵌合rna。在一些實施方案中,所述方法還包括將所述嵌合rna片段化。在一些實施方案中,所述嵌合rna的所述片段化包括在有助於所述rna部分消化的條件下使所述嵌合rna與rnase接觸。在一些實施方案中,所述方法還包括反轉錄所述嵌合rna以產生嵌合cdna。在一些實施方案中,所述方法還包括測定所述嵌合rna或嵌合cdna中源自所述嵌合rna或嵌合cdna中的每個rna的至少一部分序列。在一些實施方案中,該方法還包括鑑定存在於所述嵌合rna中的rna,從而鑑定在細胞中彼此相互作用的rna。在一些實施方案中,鑑定出細胞中至少100個、至少500個、至少1000個或多於1000個rna-rna相互作用。在一些實施方案中,鑑定出在細胞中彼此相互作用的基本上所有的rna。在一些實施方案中,鑑定出細胞中至少70%、至少80%、至少90%或超過90%的直接rna-rna相互作用。在一些實施方案中,在細胞中彼此相互作用的rna的鑑定包括使用自動測序裝置對所述嵌合rna進行序列讀段。在一些實施方案中,在細胞中彼此相互作用的rna的所述鑑定包括從所有序列讀段鑑定嵌合序列。在一些實施方案中,所述方法還包括使用計算機將嵌合rna轉化為注釋的rna簇。在一些實施方案中,所述方法還包括使用由計算機執行的統計檢驗來鑑定所述rna簇之間的直接相互作用。在一些實施方案中,所述試劑包括核酸。在一些實施方案中,所述試劑包括化學化合物。
在一些實施方案中,提供一種藥物,其中所述藥物是使用本文所述任何實施方案的方法製備的。在一些實施方案中,該方法包括將使用本文所述的任何實施方案的方法鑑定的試劑配製在藥學上可接受的載體中。在一些實施方案中,通過用於鑑定候選治療劑的方法來配製所鑑定的試劑,其中所述方法包括使用本文所述的任何實施方案的方法鑑定在細胞中彼此相互作用的rna,並評估其用於減少或增加所述rna的相互作用的能力,其中如果所述試劑能夠減少或增加所述rna的相互作用,則所述試劑是候選治療劑。在一些實施方案中,用於鑑定在細胞中彼此相互作用的rna的方法包括將rna與蛋白質交聯並將與相同蛋白質分子交聯的rna連接在一起以形成嵌合rna。在一些實施方案中,所述rna與蛋白質的交聯在完整細胞上或在細胞裂解物中進行。在一些實施方案中,所述交聯包括uv交聯。在一些實施方案中,所述方法還包括將所述蛋白質與有助於所述蛋白質在表面上固定化的試劑相關聯。在一些實施方案中,有助於固定化的所述試劑包括生物素。在一些實施方案中,該方法還包括將與相同蛋白質分子交聯的所述rna片段化。在一些實施方案中,所述片段化包括在有助於所述rna部分消化的條件下將與相同蛋白質分子交聯的所述rna與rnase接觸。在一些實施方案中,所述方法還包括將與相同蛋白質分子交聯的所述rna連接至有助於所述rna的回收的試劑。在一些實施方案中,所述連接包括將所述rna的末端連接至所述試劑。在一些實施方案中,有助於所述rna回收的所述試劑包括核酸。在一些實施方案中,所述核酸包括其上具有生物素的核酸。在一些實施方案中,所述其上具有生物素的核酸與所述rna的所述末端的所述連接包括在將與相同蛋白質分子交聯的所述rna連接在一起以形成嵌合rna之前將其上具有生物素的所述核酸連接至所述rna的5'末端。在一些實施方案中,所述方法還包括從所述嵌合rna的5'區域除去所述生物素。在一些實施方案中,所述方法還包括回收所述嵌合rna。在一些實施方案中,所述方法還包括將所述嵌合rna片段化。在一些實施方案中,所述嵌合rna的所述片段化包括在有助於所述rna部分消化的條件下使所述嵌合rna與rnase接觸。在一些實施方案中,所述方法還包括反轉錄所述嵌合rna以產生嵌合cdna。在一些實施方案中,所述方法還包括測定所述嵌合rna或嵌合cdna中的源自所述嵌合rna或嵌合cdna中的每個rna的至少一部分序列。在一些實施方案中,該方法還包括鑑定存在於所述嵌合rna中的rna,從而鑑定在細胞中彼此相互作用的rna。在一些實施方案中,鑑定出細胞中至少100個、至少500個、至少1000個或多於1000個rna-rna相互作用。在一些實施方案中,鑑定出在細胞中彼此相互作用的基本上所有的rna。在一些實施方案中,鑑定出細胞中至少70%、至少80%、至少90%或超過90%的直接rna-rna相互作用。在一些實施方案中,在細胞中彼此相互作用的rna的鑑定包括使用自動測序裝置對所述嵌合rna進行序列讀段。在一些實施方案中,在細胞中彼此相互作用的rna的所述鑑定包括從所有序列讀段鑑定嵌合序列。在一些實施方案中,所述方法還包括使用計算機將嵌合rna轉化為注釋的rna簇。在一些實施方案中,所述方法還包括使用由計算機執行的統計檢驗來鑑定所述rna簇之間的直接相互作用。在一些實施方案中,所述試劑包括核酸。在一些實施方案中,所述試劑包括化學化合物。
在一些實施方案中,提供了用於產生包含在細胞中彼此相互作用的rna的嵌合rna的方法,其中所述方法包括將rna與蛋白質中間體和/或蛋白質複合體交聯並將與蛋白質中間體和/或蛋白質複合體交聯的rna連接在一起以形成嵌合rna,並且其中所述蛋白質複合體包含兩種或更多種相互作用蛋白。在一些實施方案中,所述rna與蛋白質中間體和/或蛋白質複合體的交聯在完整細胞上或在細胞裂解物中進行。在一些實施方案中,所述交聯包括uv交聯。在一些實施方案中,所述方法還包括將所述蛋白質中間體和/或蛋白質複合體與有助於蛋白質中間體和/或蛋白質複合體在表面上固定化的試劑相關聯。在一些實施方案中,有助於固定化的所述試劑包括生物素。在一些實施方案中,所述方法還包括將與所述至少一種蛋白質分子交聯的所述rna片段化。在一些實施方案中,片段化包括在有助於所述rna的部分消化的條件下將與蛋白質中間體和/或蛋白質複合體交聯的所述rna與rnase接觸。在一些實施方案中,所述方法還包括將與蛋白質中間體和/或蛋白質複合體交聯的所述rna連接至有助於所述rna的回收的試劑。在一些實施方案中,所述連接包括將所述rna的末端連接至所述試劑。在一些實施方案中,有助於所述rna回收的所述試劑包括核酸。在一些實施方案中,所述核酸包括其上具有生物素的核酸。在一些實施方案中,其上具有生物素的所述核酸與所述rna的所述末端的連接包括在將與蛋白質中間體和/或蛋白質複合體交聯的所述rna連接在一起以形成嵌合rna之前將其上具有生物素的所述核酸連接至所述rna的5'末端。在一些實施方案中,所述方法還包括從所述嵌合rna的5'區域除去所述生物素。在一些實施方案中,所述方法還包括回收所述嵌合rna。在一些實施方案中,所述方法還包括將所述嵌合rna片段化。在一些實施方案中,所述嵌合rna的所述片段化包括在有助於所述rna部分消化的條件下使所述嵌合rna與rnase接觸。在一些實施方案中,所述方法還包括反轉錄所述嵌合rna以產生嵌合cdna。在一些實施方案中,該方法還包括鑑定存在於所述嵌合rna中的rna,從而鑑定在細胞中彼此相互作用的rna。在一些實施方案中,鑑定出細胞中至少100個、至少500個、至少1000個或多於1000個rna-rna相互作用。在一些實施方案中,鑑定出在細胞中彼此相互作用的基本上所有的rna。在一些實施方案中,鑑定出細胞中至少70%、至少80%、至少90%或超過90%的直接rna-rna相互作用。在一些實施方案中,在細胞中彼此相互作用的rna的鑑定包括使用自動測序裝置對所述嵌合rna進行序列讀段。在一些實施方案中,在細胞中彼此相互作用的rna的所述鑑定包括從所有序列讀段鑑定嵌合序列。在一些實施方案中,所述方法還包括使用計算機將嵌合rna轉化為注釋的rna簇。在一些實施方案中,所述方法還包括使用由計算機執行的統計檢驗來鑑定所述rna簇之間的直接相互作用。在一些實施方案中,在細胞中彼此相互作用的所述rna與所述蛋白質中間體或蛋白質複合體中的不同蛋白質交聯。
在一些實施方案中,提供了包含與蛋白質中間體和/或蛋白質複合體交聯的嵌合rna的分離複合體,其中所述嵌合rna包含在細胞中彼此相互作用的rna,其中所述蛋白質複合體包含兩個以上的相互作用蛋白質。在一些實施方案中,所述嵌合rna包含與所述蛋白質中間體或蛋白質複合體中的不同蛋白質交聯的rna。
本文列出的每個參考文獻通過引用整體併入本文。
參考文獻
1.engreitz,j.m.etal.rna-rnainteractionsenablespecifictargetingofnoncodingrnastonascentpre-mrnasandchromatinsites.cell159,188-199,doi:10.1016/j.cell.2014.08.018(2014).
2.ray,d.etal.acompendiumofrna-bindingmotifsfordecodinggeneregulation.nature499,172-177,doi:10.1038/nature12311(2013).
3.meister,g.argonauteproteins:functionalinsightsandemergingroles.natrevgenet14,447-459,doi:10.1038/nrg3462(2013).
4.hafner,m.etal.transcriptome-wideidentificationofrna-bindingproteinandmicrornatargetsitesbypar-clip.cell141,129-141,doi:10.1016/j.cell.2010.03.009(2010).
5.granneman,s.,kudla,g.,petfalski,e.&tollervey,d.identificationofproteinbindingsitesonu3snornaandpre-rrnabyuvcross-linkingandhigh-throughputanalysisofcdnas.proceedingsofthenationalacademyofsciencesoftheunitedstatesofamerica106,9613-9618,doi:10.1073/pnas.0901997106(2009).
6.chi,s.w.,zang,j.b.,mele,a.&darnell,r.b.argonautehits-clipdecodesmicrorna-mrnainteractionmaps.nature460,479-486,doi:10.1038/nature08170(2009).
7.helwak,a.,kudla,g.,dudnakova,t.&tollervey,d.mappingthehumanmirnainteractomebyclashrevealsfrequentnoncanonicalbinding.cell153,654-665,doi:10.1016/j.cell.2013.03.043(2013).
8.kudla,g.,granneman,s.,hahn,d.,beggs,j.d.&tollervey,d.cross-linking,ligation,andsequencingofhybridsrevealsrna-rnainteractionsinyeast.procnatlacadsciusa108,10010-10015,doi:10.1073/pnas.1017386108(2011).
9.nicolas,f.e.experimentalvalidationofmicrornatargetsusingaluciferasereportersystem.methodsinmolecularbiology732,139-152,doi:10.1007/978-1-61779-083-6_11(2011).
10.lal,a.etal.captureofmicrorna-boundmrnasidentifiesthetumorsuppressormir-34aasaregulatorofgrowthfactorsignaling.plosgenet7,e1002363,doi:10.1371/journal.pgen.1002363(2011).
11.du,t.&zamore,p.d.beginningtounderstandmicrornafunction.cellres17,661-663,doi:10.1038/cr.2007.67(2007).
12.kalhor,r.,tjong,h.,jayathilaka,n.,alber,f.&chen,l.genomearchitecturesrevealedbytetheredchromosomeconformationcaptureandpopulation-basedmodeling.naturebiotechnology30,90-98,doi:10.1038/nbt.2057(2012).
13.belton,j.m.etal.hi-c:acomprehensivetechniquetocapturetheconformationofgenomes.methods58,268-276,doi:10.1016/j.ymeth.2012.05.001(2012).
14.baigude,h.,ahsanullah,li,z.,zhou,y.&rana,t.m.mir-trap:abenchtopchemicalbiologystrategytoidentifymicrornatargets.angewchemintedengl51,5880-5883,doi:10.1002/anie.201201512(2012).
15.loeb,g.b.etal.transcriptome-widemir-155bindingmaprevealswidespreadnoncanonicalmicrornatargeting.molcell48,760-770,doi:10.1016/j.molcel.2012.10.002(2012).
16.wang,z.etal.iclippredictsthedualsplicingeffectsoftia-rnainteractions.plosbiol8,e1000530,doi:10.1371/journal.pbio.1000530(2010).
17.konig,j.etal.icliprevealsthefunctionofhnrnpparticlesinsplicingatindividualnucleotideresolution.natstructmolbiol17,909-915,doi:10.1038/nsmb.1838(2010).
18.nowak,d.e.,tian,b.&brasier,a.r.two-stepcross-linkingmethodforidentificationofnf-kappabgenenetworkbychromatinimmunoprecipitation.biotechniques39,715-725(2005).
19.zeng,p.y.,vakoc,c.r.,chen,z.c.,blobel,g.a.&berger,s.l.invivodualcross-linkingforidentificationofindirectdna-associatedproteinsbychromatinimmunoprecipitation.biotechniques41,694-698(2006).
20.zhao,j.etal.genome-wideidentificationofpolycomb-associatedrnasbyrip-seq.molcell40,939-953,doi:10.1016/j.molcel.2010.12.011(2010).
21.yu,p.etal.spatiotemporalclusteringoftheepigenomerevealsrulesofdynamicgeneregulation.genomeres23,352-364,doi:10.1101/gr.144949.112(2013).
22.ender,c.etal.ahumansnornawithmicrorna-likefunctions.molcell32,519-528,doi:10.1016/j.molcel.2008.10.017(2008).
23.brameier,m.,herwig,a.,reinhardt,r.,walter,l.&gruber,j.humanboxc/dsnornaswithmirnalikefunctions:expandingtherangeofregulatoryrnas.nucleicacidsres39,675-686,doi:10.1093/nar/gkq776(2011).
24.guttman,m.etal.chromatinsignaturerevealsoverathousandhighlyconservedlargenon-codingrnasinmammals.nature458,223-227,doi:10.1038/nature07672(2009).
25.barabasi,a.l.&oltvai,z.n.networkbiology:understandingthecell'sfunctionalorganization.natrevgenet5,101-113,doi:10.1038/nrg1272(2004).
26.shalgi,r.,pilpel,y.&oren,m.repressionoftransposable-elements-amicrornaanti-cancerdefensemechanism?trendsingenetics:tig26,253-259,doi:10.1016/j.tig.2010.03.006(2010).
27.yuan,z.,sun,x.,liu,h.&xie,j.micrornagenesderivedfromrepetitiveelementsandexpandedbysegmentalduplicationeventsinmammaliangenomes.plosone6,e17666,doi:10.1371/journal.pone.0017666(2011).
28.schwartz,s.etal.transcriptome-widemappingrevealswidespreaddynamic-regulatedpseudouridylationofncrnaandmrna.cell159,148-162,doi:10.1016/j.cell.2014.08.028(2014).
29.bellaousov,s.,reuter,j.s.,seetin,m.g.&mathews,d.h.rnastructure:webserversforrnasecondarystructurepredictionandanalysis.nucleicacidsresearch41,w471-w474,doi:doi10.1093/nar/gkt290(2013).
30.gong,c.&maquat,l.e.lncrnastransactivatestau1-mediatedmrnadecaybyduplexingwith3'utrsviaaluelements.nature470,284-288,doi:10.1038/nature09701(2011).
31.cooper,g.m.etal.distributionandintensityofconstraintinmammaliangenomicsequence.genomeres15,901-913,doi:10.1101/gr.3577405(2005).
32.kiss,t.,fayet-lebaron,e.&jady,b.e.boxh/acasmallribonucleoproteins.molcell37,597-606,doi:10.1016/j.molcel.2010.01.032(2010).
序列表
shengzhong鐘聲
tricongnguyen阮池公
rnastitchsequencing:anassayfordirectmappingofrna:rnainteractionsincells
rnastitch測序:用於直接映射細胞中rna:rna相互作用的測定
ucsd089.001wo
62/053615
2014-09-22
21
fastseq用於windows版本4.0
1
24
rna
人工序列
尚未歸類的特性
(5)...(5)
生物素化
生物素標記的rna接頭
1
cuagtagcccaugcaaugcgagga24
2
24
dna
人工序列
互補的dna寡核苷酸
尚未歸類的特性
(1)...(5)
核酸之間的硫代磷酸酯鍵
2
tcgcattgcatgggctactagcat24
3
20
dna
人工序列
3』反轉錄(rt)適配子
尚未歸類的特性
(1)...(1)
適配子4rapp
尚未歸類的特性
(20)...(20)
適配子3ddc
3
agatcggaagagcggttcag20
4
53
dna
人工序列
rt引物
尚未歸類的特性
(1)...(1)
5phos
尚未歸類的特性
(1)...(2)
n為a,c,t或g
尚未歸類的特性
(3)...(3)
n為a或c
尚未歸類的特性
(4)...(4)
n為g或a
尚未歸類的特性
(5)...(5)
n為g,c或t
尚未歸類的特性
(6)...(6)
n為t或c
4
nnnnnnnnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct53
5
10
dna
人工序列
條形碼
尚未歸類的特性
(1)...(1)
n為a,c,t或g
尚未歸類的特性
(2)...(2)
n為a,c,t或g
尚未歸類的特性
(3)...(3)
n為a,c,t或g
尚未歸類的特性
(4)...(4)
n為a,c,t或g
尚未歸類的特性
(5)...(5)
n為a或c
尚未歸類的特性
(6)...(6)
n為g或a
尚未歸類的特性
(7)...(7)
n為g,c或t
尚未歸類的特性
(8)...(8)
n為t或c
尚未歸類的特性
(9)...(9)
n為a,c,t或g
尚未歸類的特性
(10)...(10)
n為a,c,t或g
5
nnnnnnnnnn10
6
58
dna
人工序列
illuminapepcr正向引物1.0
6
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58
7
61
dna
人工序列
pepcr反向引物2.0
7
caagcagaagacggcatacgagatcggtctcggcattcctgctgaaccgctcttccgatc60
t61
8
28
dna
人工序列
切割_寡核苷酸
修飾的鹼基
(28)...(28)
idt
8
gttcaggatccacgacgctcttcaaaat28
9
19
dna
人工序列
正向引物dp5
9
cacgacgctcttccgatct19
10
20
dna
人工序列
反向引物dp3
10
ctgaaccgctcttccgatct20
11
24
rna
人工序列
生物素化的rna接頭
尚未歸類的特性
(5)...(5)
生物素化的
11
cuagtagcccaugcaaugcgagga24
12
24
dna
人工序列
含rna接頭的互補dna鏈
尚未歸類的特性
(1)...(5)
核酸鹼基之間的硫代磷酸酯鍵
12
tcgcattgcatgggctactagcat24
13
20
dna
人工序列
預腺苷化的rt適配子(無rnase的hplc-純化自idt)
尚未歸類的特性
(1)...(1)
54rapp
尚未歸類的特性
(20)...(20)
3ddc
13
agatcggaagagcggttcag20
14
52
dna
人工序列
用於es-1樣品的rt引物
尚未歸類的特性
(1)...(1)
5phos
尚未歸類的特性
(1)...(1)
n為a,c,t或g
尚未歸類的特性
(2)...(2)
n為a,c,t或g
尚未歸類的特性
(7)...(7)
n為a,c,t或g
尚未歸類的特性
(8)...(8)
n為a,c,t或g
尚未歸類的特性
(9)...(9)
n為a,c,t或g
14
nnaggtnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct52
15
53
dna
人工序列
用於es-2樣品和mef樣品的rt引物
尚未歸類的特性
(1)...(1)
5phos
尚未歸類的特性
(1)...(1)
n為a,c,t或g
尚未歸類的特性
(2)...(2)
n為a,c,t或g
尚未歸類的特性
(7)...(7)
n為a,c,t或g
尚未歸類的特性
(8)...(8)
n為a,c,t或g
尚未歸類的特性
(9)...(9)
n為a,c,t或g
尚未歸類的特性
(10)...(10)
n為a,c,t或g
15
nncgccnnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct53
16
53
dna
人工序列
用於es-間接樣品的rt引物
尚未歸類的特性
(1)...(1)
5phos
尚未歸類的特性
(1)...(1)
n為a,c,t或g
尚未歸類的特性
(2)...(2)
n為a,c,t或g
尚未歸類的特性
(7)...(7)
n為a,c,t或g
尚未歸類的特性
(8)...(8)
n為a,c,t或g
尚未歸類的特性
(9)...(9)
n為a,c,t或g
尚未歸類的特性
(10)...(10)
n為a,c,t或g
16
nncattnnnnagatcggaagagcgtcgtggatcctgaaccgctcttccgatct53
17
28
dna
人工序列
切割_寡核苷酸(hplc-純化自idt)
尚未歸類的特性
(28)...(28)
idt
17
gttcaggatccacgacgctcttcaaaat28
18
19
dna
人工序列
截短的pcr正向引物dp5
18
cacgacgctcttccgatct19
19
20
dna
人工序列
截短的pcr反向引物dp3
19
ctgaaccgctcttccgatct20
20
58
dna
人工序列
illuminapepcr正向引物1.0
20
aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct58
21
61
dna
人工序列
illuminapepcr反向引物2.0
21
caagcagaagacggcatacgagatcggtctcggcattcctgctgaaccgctcttccgatc60
t61