一種馬來酸酐熔融接枝聚烯烴材料的製備方法與流程
2023-06-11 12:11:06 7
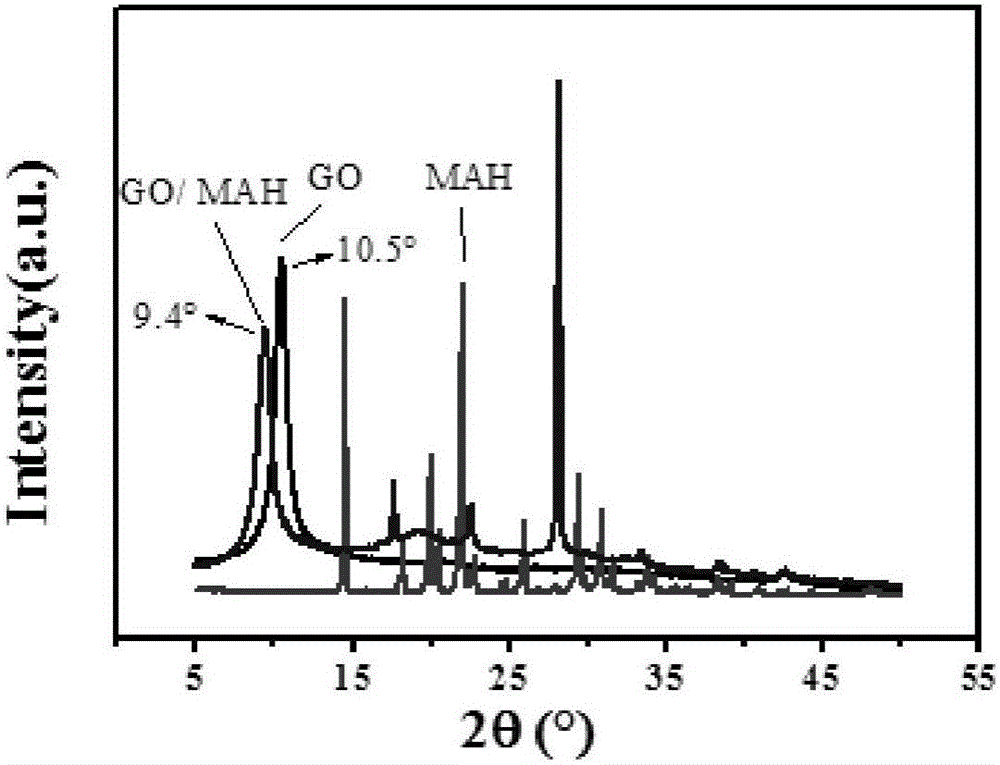
本發明屬於高分子材料領域,具體涉及高接枝率、高分子量或高接枝率、低交聯度的馬來酸酐熔融接枝聚烯烴材料的製備方法。
背景技術:
:聚烯烴(PO,polyolefin)是一種具有優異綜合性能的通用塑料,廣泛應用於汽車、電器、包裝等領域。但由於聚烯烴是非極性聚合物,與極性聚合物(尼龍、聚乳酸等)、極性填料(玻纖、碳纖等)不相容。為了提高聚烯烴的極性,通常加入過氧類引發劑和乙烯類不飽和極性單體。工業生產中,為了獲得較高的接枝率,通常要在聚烯烴中添加高含量的過氧化二異丙苯(DCP)和馬來酸酐(MAH),如添加0.5phr(重量百份數,聚烯烴含量為100份)的過氧化二異丙苯和5phr的馬來酸酐,雖然此時接枝率提高了,但由於過氧化二異丙苯含量較高,聚烯烴出現了嚴重的副反應:丙烯型聚烯烴出現降解(如聚丙烯PP),乙烯型聚烯烴出現交聯(如聚乙烯PE)。為了抑制副反應,並同時提高接枝率,通常加入苯乙烯(St)這一富電子類型的第二單體,即PO/DCP/MAH/St體系。然而該體系也存在一定的局限性,比如聚丙烯(PP,polypropylene)體系中190℃下固定馬來酸酐含量為5phr,引發劑含量較低時(≤0.3phr),苯乙烯抑制降解的作用有限,引發劑含量較高時(≥0.5phr),苯乙烯的加入反倒增加了降解,又比如聚乙烯(PE,polyethylene)體系PE/DCP/MAH/St體系主要的副反應是聚乙烯的交聯,而苯乙烯對聚乙烯交聯的抑制作用同樣有限。因此難以得到同時具有高接枝率、高分子量或高接枝率、低交聯度的馬來酸酐接枝聚烯烴PO-g-(MAH-co-St)材料。為了抑制PO/DCP/MAH/St體系的降解或交聯,本發明加入氧化石墨烯(GO)這一新型填料。熔融共混過程中,由於π-π作用(馬來酸酐上的碳碳雙鍵和氧化石墨烯上類苯環sp2雜化結構組成的大的π電子共軛體系)和氫鍵作用(馬來酸酐上的強極性羰基基團與氧化石墨烯上的羧基、羥基等極性基團),馬來酸酐能夠插入氧化石墨烯層間,使其層間距變大,從而形成氧化石墨烯-馬來酸酐複合物。該複合物能夠淬滅聚丙烯叔碳鏈段自由基(PP3·)、聚乙烯仲碳鏈段自由基(PE2·),即抑制前者發生β-斷裂,也就抑制了聚丙烯的降解,抑制後者發生交聯,也就抑制了聚乙烯的交聯。但由於苯乙烯共軛體系中豐富的電子云,PP3~St·和PE2~St·活性較高,該複合物並不能有效地淬滅這兩種連接苯乙烯的鏈段自由基,因而這兩種自由基能夠有效地進行接枝反應。這樣,較高引發劑含量下(0.3phr~3phr)聚烯烴及其衍生物體系中的降解或交聯仍能夠得到有效抑制,但PP3~St·或PE2~St·與馬來酸酐單體的接枝過程並不能被有效抑制,因而可以得到同時具有高接枝率、高分子量或高接枝率、低交聯度的馬來酸酐-苯乙烯共接枝聚烯烴材料PO-g-(MAH-co-St)。技術實現要素:本發明的目的是提供一種馬來酸酐熔融接枝聚烯烴材料的製備方法,其接枝率高、分子量高或接枝率高、交聯度低。為了實現上述目的,本發明馬來酸酐熔融接枝聚烯烴材料的製備方法,具體步驟如下:步驟(1)、將重量份數為0.01~15份的馬來酸酐、重量份數為0.01~10份的氧化石墨烯直接加入到重量份數為100份的聚烯烴粒料或粉料上,常溫下混合均勻,將該物理混合物加入到反應設備;步驟(2)、將重量份數為0.01~5份的引發劑、重量份數為0.01~10份的苯乙烯液體混合均勻,將該混合物加入反應設備,在一定溫度下熔融共混而成;其中所述的反應設備為密煉機、反應型擠出機等,反應溫度區間為160~230℃,其中密煉機轉子轉速為50~150rpm,反應型擠出機螺杆轉速為30~600rpm。所述的聚烯烴為聚丙烯、聚乙烯、以及聚丙烯衍生物和聚乙烯衍生物,其中聚丙烯衍生物為乙烯-丙烯嵌段共聚物、乙烯-丙烯無規共聚物,聚乙烯為低密度聚乙烯,高密度聚乙烯等。所述的引發劑為過氧化物類引發劑,即過氧化二異丙苯、雙叔丁基過氧異丙基苯、叔丁基過氧化氫、過氧化叔丁基苯甲酸酯、過氧化叔丁基二碳酸酯、2,5-二甲基-2,5-雙(叔丁基過氧基)己烷、、2,5-二甲基-2,5-雙(叔丁基過氧基)己炔、過氧化苯甲醯中的一種或幾種。所述的氧化石墨烯為單層或多層氧化石墨烯粉末,即純度為80%~99%,厚度為0.34~17nm,層數為1~50層,片層直徑為5~100μm,比表面積為50~1000m2/g,褐黃色或黑色粉末,表面及邊緣主要為羧基、羥基、環氧基團,可噸級量產的單層或多層氧化石墨烯。與現有技術相比,本發明的優點和有益效果如下:(1)本發明製備的馬來酸酐接枝聚烯烴接枝率高、分子量高,或是接枝率高、交聯度低;(2)本發明與馬來酸酐-苯乙烯共單體接枝聚烯烴體系相比,由於氧化石墨烯的加入可有效抑制聚丙烯叔碳鏈段自由基的降解、聚乙烯仲碳鏈段自由基的交聯,較高的引發劑含量並不會引起較大降解或交聯,可實現降解和交聯的有效控制或調節;(3)本發明首次將氧化石墨烯應用於馬來酸酐熔融接枝聚烯烴體系中,不同於以往氧化石墨烯在複合材料、藥物負載等體系的應用,本發明拓展了氧化石墨烯的應用範圍,為氧化石墨烯的大規模應用提供了新的可能。(4)本發明製備的馬來酸酐接枝聚烯烴不同於普通的增容劑,該增容劑中存在氧化石墨烯,具有較高的熱穩定性、氣體阻隔性能等;(5)本發明在馬來酸酐接枝聚烯烴增容劑製備過程中完全不涉及溶劑的使用,所有組分並不需要進一步處理,直接通過攪拌機進行物理混合;(6)本發明通過密煉機或擠出機熔融共混製備所需增容劑,該方法具有工業大規模生產的可能。附圖說明圖1為GO、GO-MAH、MAH的紅外圖譜;圖2為GO、GO-MAH、MAH的X射線衍射圖譜;圖3為GO、GO-MAH、MAH的熱失重穩定曲線的重量-溫度圖譜;圖4為GO、GO-MAH、MAH的熱失重穩定曲線的失重速率-溫度圖譜;圖5為製備得到的實施例1和比較例1樣品照片。具體實施方式下一步結合實施例,進一步說明本發明:實施例中為了測定馬來酸酐接枝聚烯烴的接枝率,首先對PO-g-MAH接枝物進行純化,然後採用紅外光譜和酸鹼滴定結合的測定方法來表徵接枝率。PO-g-MAH純化的具體步驟如下:首先,取1.5g樣品加入50ml二甲苯中,140℃油浴30min,樣品完全溶解,趁熱將溶液倒入100ml丙酮中,然後將該濾液抽濾,抽濾物70℃真空乾燥3h;其次,將乾燥過的抽濾物用濾紙包裹起來放入丙酮抽提裝置,90℃油浴下抽提24h;最後,將抽提物70℃真空乾燥8h,得到乾燥的純化樣品。傅立葉紅外光譜法(FT-IR)的具體步驟如下:首先,取0.3g左右純化樣品置於聚四氟乙烯膜上,平板硫化儀中190℃下預熱5min,60MPa下熱壓2min,通水冷卻至常溫取出,輕輕將所壓薄膜揭起;其次,選擇100μm厚度左右的薄膜進行紅外測試:透過模式TR,吸光度absorbance,解析度1cm-1,掃描範圍:4000-400cm-1,掃描次數64scans;最後,使用omnic軟體分別對1780cm-1、2721cm-1、1470cm-1峰進行積分,得到峰面積A1780、A2721和A1470,A1780/A2721、A1780/A1470即為吸光度之比Ra,可定量來表徵接枝率的大小。其中1780cm-1為酸酐中的羰基峰,2721cm-1為聚丙烯鏈段骨架峰,1470cm-1為聚乙烯亞甲基特徵峰,即內標峰。酸鹼滴定純化樣品的具體步驟如下:首先,取0.5g左右純化樣品放入裝有50ml二甲苯的三頸燒瓶(容積250ml)中,油浴溫度150℃,加熱回流20min,樣品充分溶解;其次,將油浴溫度調至100℃,並使三頸燒瓶懸空於油浴裝置,待三頸燒瓶中二甲苯溶液冷卻至70℃,磁子攪拌溶液的同時緩慢滴入10ml氫氧化鉀-異丙醇溶液,此時將三頸燒瓶置於油浴中100℃加熱回流1.5h,使羧基化的馬來酸酐與氫氧化鉀充分反應;然後,冷卻油浴至80℃,此時溶液溫度70℃,趁熱滴加5滴酚酞溶液,溶液呈粉紅色;最後用鹽酸-異丙醇溶液趁熱滴定至無色,該過程中由於滴入的鹽酸-異丙醇溶液是常溫的,所以滴定過程中的二甲苯溶液溫度會降低,可在滴定過程中適當升高油浴溫度來保持二甲苯溶液溫度為70℃。重複該實驗3次,計算出接枝率G1。空白樣(不加純化樣品)的滴定過程同上,計算出接枝率G2。最終,酸鹼滴定法得到的純化樣品的接枝率為,G=G1-G2。熔融指數測定按照GB3682-83標準在高鐵熔融指數儀上進行測試:230℃,2.16kg。下面結合實施例對本發明進行詳細說明。然而,這些實施例僅作為示例性目的,本發明的範圍不限於此。實施例1:PP/DCP/MAH/St/GOPP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例1:PP/DCP/MAH/StPP/DCP/MAH按照重量比100/0.5/5(其中聚丙烯稱量46g)進行混合併進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例2:PP/DCP/MAH/GOPP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例3:PP/DCP/St/GOPP/DCP/GO按照重量比100/0.5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例4:PP/DCP/MAHPP/DCP/MAH按照重量比100/0.5/5(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例5:PP/DCP/StPP/DCP按照重量比100/0.5(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例6:PP/DCP/GOPP/DCP/GO按照重量比100/0.5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例7:PP/DCPPP/DCP按照重量比100/0.5(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例8:純PP按照46g聚丙烯固體顆粒加入密煉機中,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。實施例2:PP/DCP/MAH/St/GO(改變DCP含量為1.0phr)PP/DCP/MAH按照重量比GO/100/1.0/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例9:PP/DCP/MAH/GO/100/0.5/5/0.1PP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。實施例3:PP/DCP/MAH/St/GO(改變溫度為180℃)PP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,180℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例10:PP/DCP/MAH/100/0.5/5,180℃PP/DCP/MAH按照重量比100/0.5/5(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,180℃下以50rpm的速度混煉5min。樣品的測試結果見表1。比較例11:PP/DCP/MAH/GO/100/0.5/5/0.1PP/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。實施例4:PP/DCP/MAH/St/GO,180℃PP/DCP/MAH/GO按照重量比100/3.0/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,180℃下以50rpm的速度混煉5min。樣品的測試結果見表1。實施例5:PP/DCP/MAH/St/GOPP/DCP/MAH/GO按照重量比100/3.0/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表1。實施例6:PP-g-MAH的熱失重穩定性。可以看出,PP/DCP/MAH/100/0.5/5(190℃)中加入0.1phr的GO之後,T5%提高了11℃,Tmax降低了2.5℃。樣品的測試結果見表2。實施例7:PE/DCP/MAH/St/GOPE/DCP/MAH/GO按照重量比100/0.5/5/0.1(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表3。比較例12:PE/DCP/MAH/StPE/DCP/MAH按照重量比100/0.5/5(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,而後再加入2.5phr的液體狀St,190℃下以50rpm的速度混煉5min。樣品的測試結果見表3。比較例13:PE/DCP/MAHPE/DCP/MAH按照重量比100/0.5/5(其中聚丙烯稱量46g)進行混合併手動攪拌均勻,加入密煉機中,190℃下以50rpm的速度混煉5min。樣品的測試結果見表3。實施例8:GO、GO-MAH、MAH的紅外圖譜。詳見圖1。其中GO-MAH紅外樣品製備方法如下:分別稱量39mg的GO粉末、28mg的塊狀MAH,使用研缽研磨,研磨過程中滴加3滴丙酮;40℃真空乾燥12h後,該複合物出現塊狀物,重新研磨。GO、MAH兩個樣品與GO-MAH樣品經歷了相同的製備過程,研磨過程中滴加丙酮,然後乾燥。選取少量的乾燥樣品,使用溴化鉀壓片法壓片,透射模式,對製備樣品進行紅外測試(經過對比,未經處理與研磨-乾燥處理的的GO、MAH樣品的紅外圖譜相同,排除了研磨-乾燥處理過程對GO、MAH樣品的影響)首先,相比於GO,GO/MAH的變化如下,(1)GO/MAH中的羥基「OH「峰向低波數偏移,且峰形鈍化,這可能是由於氫鍵的存在(酸酐與GO上的羧基、羥基之間);(2)GO中存在大的吸收峰:3683-2814cm-1,GO/MAH中的對應吸收峰範圍變大很多:3683-1860cm-1,這可能是由於GO中的不飽和六元碳環π-π共軛結構與MAH中碳碳雙鍵π-π共軛結構發生共軛,從而使1780cm-1、1860cm-1等羰基吸收峰消失,3000-2000cm-1範圍的碳氫峰波長範圍變大;(3)羰基峰略向高波數偏移,1706cm-1到1717cm-1,且強度相對碳碳雙鍵變弱,可能是由於酸酐中的高波數羰基吸收峰的影響,即與GO中的羰基發生氫鍵作用,因而向高波數偏移,同時由於GO與MAH間的共軛作用,羰基峰強度相對碳碳雙鍵變弱;(4)「C-OH」峰消失,可能是由於MAH與其反應;其次,相比於MAH,GO/MAH的變化如下,GO-MAH中1780-1860cm-1的羰基伸縮振動峰消失,只剩下1717cm-1的羰基吸收峰,這可能是由於GO與MAH之間的共軛效應、氫鍵效應,使其電子云密度平均化。樣品的測試結果見圖1。實施例9:X射線衍射,GO、GO/MAH、MAH的XRD樣品使用實施例8中紅外製備樣品。詳見圖2。根據布拉格方程2dsinθ=nλ計算可知,氧化石墨烯與馬來酸酐研磨以後,層間距由0.77nm變為0.94nm,這表明馬來酸酐插入氧化石墨烯層間,表明二者間由明顯的相互作用,與紅外結果對應,表明熔融接枝過程中確實生成了GO/MAH複合物,正是該複合物抑制了其降解、接枝。樣品的測試結果見圖2。實施例10:熱失重穩定性曲線,GO、GO/MAH、MAH的XRD樣品使用實施例8中紅外製備樣品。GO-MAH的熱穩定性下降,GO-MAH在100℃附近出現新的Tmax特徵峰,這應當是由於MAH插入了GO層間,並且插入GO層間的MAH的熱穩定性低於純的MAH,這表明馬來酸酐插入氧化石墨烯層間,表明二者間由明顯的相互作用。樣品的測試結果見表1、圖3和圖4。實施例11:PP-g-MAH(實施例1和比較例1)樣品的圖片和SEM圖。圖5中可以看出,片狀的比較例1樣品為乳白色,而片狀的實施例1樣品為灰色,雖然有少量氧化石墨烯發生團聚形成黑色小顆粒,但總的來說氧化石墨烯還是均勻分散於其中的。樣品的測試結果見表1和圖5。表1.馬來酸酐接枝聚丙烯體系的熔指和吸光度之比SampleT5%(℃)Tmax(℃)NeatPP355426.4PP/DCP/MAH/100/0.5/5(190℃)398468.2PP/DCP/MAH/GO/100/0.5/5/0.1(190℃)409465.7PP/DCP/MAH/GO/100/0.5/5/0.1(190℃)412466.1表2.PP-g-MAH的熱失重穩定性表3.馬來酸酐接枝聚乙烯體系的熔指和吸光度之比實施例12:將5gMAH、0.05gGO直接加入到50g乙烯-丙烯嵌段共聚物粒料中,將該混合物攪拌均勻後加入到密煉機;將0.005g雙叔丁基過氧異丙基苯、0.005gSt混勻後加入到上述密煉機中,160℃下以100rpm的速度混煉5min。實施例13:將0.005gMAH、0.5gGO直接加入到50g乙烯-丙烯嵌段共聚物粒料中,將該混合物攪拌均勻後加入到密煉機;將2.5g叔丁基過氧化氫、0.5gSt混勻後加入到上述密煉機中,230℃下以60rpm的速度混煉5min。實施例14:將7.5gMAH、1gGO直接加入到50g等規聚丙烯粒料中,將該混合物攪拌均勻後加入到螺杆擠出機;將1g過氧化叔丁基苯甲酸酯、2.5gSt混勻後加入到上述螺杆擠出機中,180℃下以200rpm的速度擠出。實施例15:將0.05gMAH、5gGO直接加入到50g乙烯-丙烯無規共聚物粒料中,將該混合物攪拌均勻後加入到螺杆擠出機;將0.05g過氧化叔丁基苯二碳酸酯、5gSt混勻後加入到上述螺杆擠出機中,170℃下以400rpm的速度擠出。實施例16:將500gMAH、5gGO直接加入到5000g高密度聚乙烯粒料中,將該混合物攪拌均勻後加入到螺杆擠出機;將5g過氧化叔丁基苯二碳酸酯、500gSt混勻後加入到上述螺杆擠出機中,170℃下以400rpm的速度擠出。實施例17:將50gMAH、1gGO直接加入到5000g高密度聚乙烯粒料中,將該混合物攪拌均勻後加入到螺杆擠出機;將0.2g過氧化叔丁基苯二碳酸酯、0.3g過氧化叔丁基苯甲酸酯、50gSt混勻後加入到上述螺杆擠出機中,230℃下以100rpm的速度擠出。上述實施例並非是對於本發明的限制,本發明並非僅限於上述實施例,只要符合本發明要求,均屬於本發明的保護範圍。當前第1頁1 2 3