核酸序列的分析的製作方法
2023-06-03 02:58:11 7
本申請要求2014年6月26日提交的美國臨時專利申請號62/017,808以及2014年10月29日提交的美國臨時專利申請號62/072,214的優先權,這些申請中的每一者出於所有目的以全文引用的方式併入本文中。
背景
對特定人基因組的基本理解可能不只需要簡單識別存在或不存在某些基因變異,諸如突變。確定某些基因變異是出現在同一染色體還是不同染色體上(也稱為定相(phasing))也是重要的。關於基因變異模式(諸如單倍型)的信息也是重要的,關於基因拷貝的數目的信息也重要。
術語「單倍型」是指在連續區塊(block)中一起遺傳的DNA序列變體(等位基因)的集合。一般來說,人基因組含有各基因的兩個拷貝–母體拷貝和父體拷貝。對於各自具有兩個可能的等位基因的一對基因,例如基因等位基因「A」和「a」,以及基因等位基因「B」和「b」,給定個體的基因組將包括兩種單倍型中的一種:「AB/ab」,其中A和B等位基因位於同一染色體上(「順式」構型);或「Ab/aB,其中A和B等位基因位於不同染色體上(「反式」構型)。可使用定相方法或分析來確定指定等位基因集合是位於同一染色體還是不同染色體上。在一些情況下,限定單倍型的若干相連等位基因可能與特定疾病表型相關聯或相關;在此類情況下,就患者是否將展示疾病來說單倍型(而不是任何一種特定基因變體)可能是最具決定性的因素。
基因拷貝數也在一些疾病表型中發揮作用。大多數基因通常以兩個拷貝存在,然而,擴增的基因為以超過兩個功能拷貝的形式存在的基因。在一些情況下,基因還可經歷功能拷貝減少。基因拷貝數的減少或增加可導致異常mRNA和蛋白質表達水平的產生,從而潛在地導致癌性狀態或其他病症。癌症和其他基因病症經常與異常(增加或減少)的染色體數目(「非整倍性」)相關聯。可使用諸如螢光原位雜交或比較基因組雜交等細胞遺傳傳技術來檢測異常基因的存在或染色體拷貝數。本領域中需要改進的檢測基因定相信息、單倍型或拷貝數變異的方法。
概述
本公開提供可適用於在基因材料的表徵方面提供顯著進步的方法和系統。這些方法和系統可適用於提供使用通常可獲得的技術可能大體上較難實現的基因表徵,包括例如單倍型定相、識別結構變異(例如缺失、重複、拷貝數變體、插入、倒位、易位、長串聯重複(LTR)、短串聯重複(STR))以及多種其他適用表徵。
本公開一個方面提供一種用於識別核酸中的一種或多種變異的方法,其包括:a)提供核酸的第一片段,其中第一片段具有大於10千鹼基(kb)的長度;(b)對第一片段的多個第二片段進行測序以提供多個片段序列,所述多個片段序列共有共同的條形碼序列;(c)根據共同條形碼序列的存在將多個片段序列歸屬於第一片段;(d)使用多個片段序列測定第一片段的核酸序列,其中以低於1%的誤差率測定核酸序列;以及(e)識別(d)中所測定的第一片段的核酸序列中的一種或多種變異,由此識別核酸內的一種或多種變異。
在一些情況下,第一片段在多個離散分區之中的離散分區中。在一些情況下,離散分區為乳液中的微滴。在一些情況下,識別包括識別第一片段的核酸序列中的定相變體。在一些情況下,識別包括由第一片段的核酸序列識別核酸中的一種或多種結構變異。在一些情況下,第一片段具有大於15kb的長度。在一些情況下,第一片段具有大於20kb的長度。在一些情況下,測定包括將多個片段序列映射至參考序列。在一些情況下,測定包括組裝具有共同條形碼序列的多個片段序列。
在一些情況下,用於識別一種或多種變異的方法進一步包括提供核酸的多個第一片段,其長度為至少10kb,並且識別包括測定多個第一片段中的每一者的核酸序列以及由多個第一片段中的每一者的核酸序列識別核酸中的一種或多種變異。
在一些情況下,用於識別一種或多種變異的方法進一步包括基於多個第一片段的兩個或更多個核酸序列的重疊核酸序列將兩個或更多個核酸序列連接成推測重疊群(inferred contig),其中最大推測重疊群長度為至少10kb。在一些情況下,最大推測重疊群長度為至少20kb。在一些情況下,最大推測重疊群長度為至少40kb。在一些情況下,最大推測重疊群長度為至少50kb。在一些情況下,最大推測重疊群長度為至少100kb。在一些情況下,最大推測重疊群長度為至少200kb。在一些情況下,最大推測重疊群長度為至少500kb。在一些情況下,最大推測重疊群長度為至少750kb。在一些情況下,最大推測重疊群長度為至少1兆鹼基(Mb)。在一些情況下,最大推測重疊群長度為至少1.75Mb。在一些情況下,最大推測重疊群長度為至少2.5Mb。
在一些情況下,識別一種或多種變異的方法進一步包括基於多個第一片段的兩個或更多個核酸序列內的重疊定相變體將多個第一片段的兩個或更多個核酸序列連接成相位區塊(phase block),其中最大相位區塊長度為至少10kb。在一些情況下,最大相位區塊長度為至少20kb。在一些情況下,最大相位區塊長度為至少40kb。在一些情況下,最大相位區塊長度為至少50kb。在一些情況下,最大相位區塊長度為至少100kb。在一些情況下,最大相位區塊長度為至少200kb。在一些情況下,最大相位區塊長度為至少500kb。在一些情況下,最大相位區塊長度為至少750kb。在一些情況下,最大相位區塊長度為至少1Mb。在一些情況下,最大相位區塊長度為至少1.75Mb。在一些情況下,最大相位區塊長度為至少2.5Mb。
在一些情況下,用於識別一種或多種變異的方法進一步包括基於多個第一片段的兩個或更多個核酸序列的重疊核酸序列將兩個或更多個核酸序列連接成推測重疊群,由此形成推測重疊群的群體,其中推測重疊群的群體的N50為至少10kb。在一些情況下,推測重疊群的群體的N50為至少20kb。在一些情況下,推測重疊群的群體的N50為至少40kb。在一些情況下,推測重疊群的群體的N50為至少50kb。在一些情況下,推測重疊群的群體的N50為至少100kb。在一些情況下,推測重疊群的群體的N50為至少200kb。在一些情況下,推測重疊群的群體的N50為至少500kb。在一些情況下,推測重疊群的群體的N50為至少750kb。在一些情況下,推測重疊群的群體的N50為至少1Mb。在一些情況下,推測重疊群的群體的N50為至少1.75Mb。在一些情況下,推測重疊群的群體的N50為至少2.5Mb。
在一些情況下,用於識別一種或多種變異的方法進一步包括基於多個第一片段的兩個或更多個核酸序列內的重疊定相變體將多個第一片段的兩個或更多個核酸序列連接成相位區塊,由此形成相位區塊的群體,其中相位區塊的群體的N50為至少10kb。在一些情況下,相位區塊的群體的N50為至少20kb。在一些情況下,相位區塊的群體的N50為至少40kb。在一些情況下,相位區塊的群體的N50為至少50kb。在一些情況下,相位區塊的群體的N50為至少100kb。在一些情況下,相位區塊的群體的N50為至少200kb。在一些情況下,相位區塊的群體的N50為至少500kb。在一些情況下,相位區塊的群體的N50為至少750kb。在一些情況下,相位區塊的群體的N50為至少1Mb。在一些情況下,相位區塊的群體的N50為至少1.75Mb。在一些情況下,相位區塊的群體的N50為至少2.5Mb。
本公開的額外方面提供一種用於確定核酸結構變異的存在的方法。所述方法可包括:(a)提供核酸的多個第一片段分子,其中多個第一片段分子中的給定第一片段分子包含結構變異;(b)對多個第一片段分子中的每一者的多個第二片段分子進行測序以提供多個片段序列,其中多個片段序列中對應於給定第一片段分子的每一者共有共同的條形碼序列;以及(c)通過以下方式確定結構變異的存在:(i)將多個片段序列映射至參考序列,(ii)識別共有共同的條形碼序列的多個片段序列,並且(iii)基於在比給定第一片段分子的長度相隔更遠的位置存在升高量的映射至參考序列的共有共同條形碼序列的多個片段序列來識別結構變異,所述升高量是相對於缺少結構變異的序列。
在一些情況下,升高量相對於源於核酸中具有結構變異的區域的第一片段分子的總數為1%或更多。在一些情況下,升高量相對於源於核酸中具有結構變異的區域的第一片段分子的總數為2%或更多。在一些情況下,所述位置相隔至少約100個鹼基。在一些情況下,所述位置相隔至少約500個鹼基。在一些情況下,所述位置相隔至少約1千鹼基(kb)。在一些情況下,所述位置相隔至少約10kb。
在一些情況下,確定核酸的結構變異的存在的方法進一步包括通過由多個片段序列形成給定第一片段分子的組裝物(assembly)來識別結構變異,其中基於共同條形碼序列的存在選擇多個片段序列作為組裝物的輸入物(input)。在一些情況下,組裝物通過由多個片段序列產生共有序列來形成。在一些情況下,結構變異包括易位。
本公開的額外方面提供一種表徵變體核酸序列的方法。在一些情況下,所述方法可包括:(a)將變體核酸片段化以提供具有大於10千鹼基(kb)的長度的多個第一片段;(b)將多個第一片段分離至離散分區中;(c)從各個第一片段各自的分區內的各個第一片段形成多個第二片段,所述多個第二片段具有與其連接的條形碼序列,給定分區內的所述條形碼序列為共同條形碼序列;(d)對多個第二片段和與其連接的條形碼序列進行測序,以提供多個第二片段序列;(e)至少部分基於共同條形碼序列的存在將第二片段序列歸屬於原始第一片段,以提供第二片段序列的第一片段序列環境(context);以及(f)由第一片段序列環境識別變體核酸的變體部分,由此表徵變體核酸序列。在一些情況下,歸屬包括至少部分基於共同條形碼序列的存在由多個第二片段序列組裝多個第一片段中的單個片段的序列的至少一部分。在一些情況下,歸屬包括至少部分基於共同條形碼序列將多個第二片段序列映射至多個第一片段中的單個第一片段。
在一些情況下,表徵變體核酸序列的方法進一步包括基於多個第一片段中的兩者或更多者之間的重疊序列將多個第一片段中的兩者或更多者連接成推測重疊群。在一些情況下,識別包括由第一片段序列環境識別一個或多個定相變體。在一些情況下,表徵變體核酸序列的方法進一步包括基於多個第一片段中的兩者或更多者之間的重疊定相變體將多個第一片段中的兩者或更多者連接成相位區塊。在一些情況下,識別包括由第一片段序列環境識別一種或多種結構變異。在一些情況下,一種或多種結構變異獨立地選自插入、缺失、易位、反轉錄轉座子、倒位以及重複。在一些情況下,結構變異包括插入或易位,並且第一片段序列環境指示插入或易位的存在。
本公開的額外方面提供一種識別核酸序列中的變體的方法。在一些情況下,所述方法包括:獲得核酸的多個單個片段分子的核酸序列,多個單個片段分子的核酸序列各自具有至少1千鹼基(kb)的長度;將多個單個片段分子中的一者或多者的序列連接成一個或多個推測重疊群;以及由一個或多個推測重疊群識別一個或多個變體。在一些情況下,獲得包括獲得長度大於10kb的多個片段分子的核酸序列。在一些情況下,獲得包括:提供多個單個片段分子中的各個單個片段分子的多個條形碼化片段,給定單個片段分子的條形碼化片段具有共同條形碼;對多個單個片段分子的多個條形碼化片段進行測序,測序提供低於1%的測序誤差率;以及由多個條形碼化片段和其相關條形碼的序列測定多個單個片段分子的序列。
在一些情況下,連接包括識別兩個或更多個單個片段分子之間的一個或多個重疊序列以將兩個或更多個單個片段分子連接成一個或多個推測重疊群。在一些情況下,連接包括識別兩個或更多個單個片段分子之間的一個或多個共同變體以將兩個或更多個單個片段分子連接成一個或多個推測重疊群。在一些情況下,一個或多個共同變體為定相變體,並且一個或多個推測重疊群包含至少100kb的最大相位區塊長度。在一些情況下,所述識別中所識別的一個或多個變體包含結構變異。在一些情況下,結構變異選自插入、缺失、易位、反轉錄轉座子、倒位以及重複。
本公開的額外方面提供一種表徵核酸的方法。在一些情況下,所述方法包括:獲得具有至少10千鹼基(kb)的長度的多個片段分子的核酸序列;識別多個片段分子的核酸序列中的一個或多個定相變體位置;基於第一和第二片段分子內的一個或多個共同定相變體位置的存在將至少一個第一片段分子的核酸序列連接至至少一個第二片段分子,以提供具有至少10kb的最大相位區塊長度的相位區塊;以及由具有至少10kb的最大相位區塊長度的相位區塊識別一個或多個定相變體。在一些情況下,表徵核酸的方法進一步包括由相位區塊識別一個或多個額外定相變體。在一些情況下,多個片段分子在離散分區中。在一些情況下,離散分區為乳液中的微滴。在一些情況下,多個片段分子的長度為至少50kb。在一些情況下,多個片段分子的長度為至少100kb。在一些情況下,最大相位區塊長度為至少50kb。在一些情況下,最大相位區塊長度為至少100kb。在一些情況下,最大相位區塊長度為至少1Mb。在一些情況下,最大相位區塊長度為至少2Mb。在一些情況下,最大相位區塊長度為至少2.5Mb。
本公開的額外方面提供一種方法,其包括:(a)將第一核酸分配至第一分區中,其中第一核酸包含源於生物體的第一染色體的靶序列;(b)將第二核酸分配至第二分區中,其中第二核酸包含源於生物體的第二染色體的靶序列;(c)在第一分區中,將第一條形碼序列連接至第一核酸的片段或第一核酸的諸多個部分的拷貝以提供第一條形碼化片段;(d)在第二分區中,將第二條形碼序列連接至第二核酸的片段或第二核酸的諸多個部分的拷貝以提供第二條形碼化片段,第二條形碼序列不同於第一條形碼序列;(e)測定第一和第二條形碼化片段的核酸序列,並且組裝第一和第二核酸的核酸序列;以及(f)比較第一和第二核酸的核酸序列,以分別將第一和第二核酸表徵為源於第一和第二染色體。在一些情況下,將包含第一條形碼序列的寡核苷酸與第一核酸共分配,並且將包含第二條形碼序列的寡核苷酸與第二核酸共分配。在一些情況下,將包含第一條形碼序列的寡核苷酸可釋放地連接至第一珠粒,並且將包含第二條形碼序列的寡核苷酸可釋放地連接至第二珠粒,並且共分配包括分別將第一和第二珠粒共分配至第一和第二分區中。在一些情況下,第一和第二分區包括乳液中的微滴。在一些情況下,第一染色體為父體染色體,並且第二染色體為母體染色體。在一些情況下,第一染色體和第二染色體為同源染色體。在一些情況下,第一核酸和第二核酸包含一種或多種變異。
在一些情況下,第一和第二染色體源於胎兒。在一些情況下,第一和第二核酸從取自孕婦的樣品獲得。在一些情況下,第一染色體為染色體21、18或13。在一些情況下,第二染色體為染色體21、18或13。在一些情況下,所述方法進一步包括測定第一或第二染色體的相對量。在一些情況下,所述方法進一步包括測定第一或第二染色體相對於參考染色體的量。在一些情況下,第一染色體或第二染色體或兩者的拷貝數增加。在一些情況下,拷貝數增加是癌症或非整倍性的結果。在一些情況下,第一染色體或第二染色體或兩者的拷貝數減少。在一些情況下,拷貝數減少是癌症或非整倍性的結果。
本公開的額外方面提供一種方法,其包括:(a)將第一核酸分配至第一分區中,其中第一核酸包含源於生物體的第一染色體的靶序列;(b)將第二核酸分配至第二分區中,其中第二核酸包含源於生物體的第二染色體的靶序列;(c)在第一分區中,將第一條形碼序列連接至第一核酸的片段或第一核酸的諸多個部分的拷貝以提供第一條形碼化片段;(d)在第二分區中,將第二條形碼序列連接至第二核酸的片段或第二核酸的諸多個部分的拷貝以提供第二條形碼化片段,第二條形碼序列不同於第一條形碼序列;(e)測定第一和第二條形碼化片段的核酸序列,並且組裝第一和第二核酸的核酸序列;以及(f)比較第一和第二核酸的核酸序列,以識別第一和第二核酸的核酸序列之間的任何變異。在一些情況下,將包含第一條形碼序列的寡核苷酸與第一核酸共分配,並且將包含第二條形碼序列的寡核苷酸與第二核酸共分配。在一些情況下,將包含第一條形碼序列的寡核苷酸可釋放地連接至第一珠粒,並且將包含第二條形碼序列的寡核苷酸可釋放地連接至第二珠粒,並且共分配包括分別將第一和第二珠粒共分配至第一和第二分區中。在一些情況下,第一和第二分區包括乳液中的微滴。在一些情況下,第一染色體為父體染色體,並且第二染色體為母體染色體。在一些情況下,第一染色體和第二染色體為同源染色體。在一些情況下,第一核酸和第二核酸包含一種或多種變異。在一些情況下,第一和第二染色體源於胎兒。在一些情況下,第一和第二核酸從取自孕婦的樣品獲得。在一些情況下,第一染色體為染色體21、18或13。在一些情況下,第二染色體為染色體21、18或13。在一些情況下,所述方法進一步包括測定第一或第二染色體的相對量。在一些情況下,所述方法進一步包括測定第一或第二染色體相對於參考染色體的量。在一些情況下,第一染色體或第二染色體或兩者的拷貝數增加。在一些情況下,拷貝數增加是癌症或非整倍性的結果。在一些情況下,第一染色體或第二染色體或兩者的拷貝數減少。在一些情況下,拷貝數減少是癌症或非整倍性的結果。
本公開的額外方面提供一種用於表徵胎兒核酸序列的方法。在一些情況下,所述方法包括:(a)通過以下方式測定母體核酸序列,其中母體核酸源於胎兒的懷孕母親:(i)將母體核酸片段化以提供多個第一母體片段;(ii)將多個第一母體片段分離至母體分區中;(iii)從第一母體片段各自的母體分區內的第一母體片段中的每一者形成多個第二母體片段,多個第二母體片段具有與其連接的第一條形碼序列,其中在母體分區中的給定母體分區內,第二母體片段包含與其連接的第一共同條形碼序列;(iv)對多個第二母體片段進行測序以提供多個母體片段序列;(v)至少部分基於第一共同條形碼序列的存在將母體片段序列歸屬於原始第一母體片段以測定母體核酸序列;(b)通過以下方式測定父體核酸序列,其中父體核酸源於胎兒的父親:(i)將父體核酸片段化以提供多個第一父體片段;(ii)將多個第一父體片段分離至父體離散分區中;(iii)從各個第一父體片段各自的分區內的各個第一父體片段形成多個第二父體片段,多個第二父體片段具有與其連接的第二條形碼序列,其中在給定父體分區內,第二父體片段包含與其連接的第二共同條形碼序列;(iv)對多個第二父體片段和與其連接的第二條形碼序列進行測序,以提供多個父體片段序列;(v)至少部分基於第二共同條形碼序列的存在將父體片段序列歸屬於原始第一父體片段以測定父體核酸序列;(c)從懷孕母親獲得胎兒核酸並且使用母體核酸序列和父體核酸序列測定胎兒核酸的序列和/或胎兒核酸的序列的一種或多種基因變異。
在一些情況下,使用父體片段序列和母體片段序列各者來將序列連接成一個或多個推測重疊群。在一些情況下,使用推測重疊群來構建母體和父體相位區塊。在一些情況下,將胎兒核酸的序列與母體和父體相位區塊相比較以構建胎兒相位區塊。在一些情況下,組裝父體片段序列以產生多個第一父體片段的序列的至少一部分,由此測定父體核酸序列,並且其中組裝母體片段序列以產生多個第一母體片段的序列的至少一部分,由此測定母體核酸序列。在一些情況下,測定父體核酸序列包括將父體片段序列映射至父體參考序列,並且其中測定母體核酸序列包括將母體片段序列映射至母體參考序列。
在一些情況下,以至少99%的準確度測定胎兒核酸的序列。在一些情況下,以至少99%的準確度測定胎兒核酸的序列的一種或多種基因變異。在一些情況下,一種或多種基因變異選自結構變異和單核苷酸多態性(SNP)。在一些情況下,一種或多種基因變異為選自拷貝數變異、插入、缺失、易位、反轉錄轉座子、倒位、重排、重複擴增以及重複的結構變異。
在一些情況下,用於表徵胎兒核酸序列的方法進一步包括,在(c)中,使用針對母體核酸序列和父體核酸序列所測定的一種或多種基因變異測定胎兒核酸的序列的一種或多種基因變異。在一些情況下,用於表徵胎兒核酸序列的方法進一步包括在(c)中,測定胎兒核酸的一個或多個從頭突變。在一些情況下,用於表徵胎兒核酸序列的方法進一步包括,在(c)期間或之後,測定與胎兒核酸相關的非整倍性。
在一些情況下,用於表徵胎兒核酸序列的方法進一步包括,在(a)中在(v)期間或之後,對母體核酸序列進行單倍型分析以提供單倍型解析型母體核酸序列,以及在(b)中在(v)期間或之後,對父體核酸序列進行單倍型分析以提供單倍型解析型父體核酸序列。在一些情況下,用於表徵胎兒核酸序列的方法進一步包括在(c)中,使用單倍型解析型母體核酸序列和單倍型解析型父體核酸序列測定胎兒核酸的序列和/或一種或多種基因變異。在一些情況下,母體核酸和父體核酸中的一者或多者為基因組脫氧核糖核酸(DNA)。在一些情況下,在(c)中,胎兒核酸包括無細胞核酸。在一些情況下,用於表徵胎兒核酸序列的方法進一步包括,在(a)中,以至少99%的準確度測定母體核酸序列。在一些情況下,表徵胎兒核酸序列的方法進一步包括,在(b)中,以至少99%的準確度測定父體核酸序列。
在一些情況下,母體核酸序列和/或父體核酸序列具有大於10千鹼基(kb)的長度。在一些情況下,母體和父體分區包括乳液中的微滴。在一些情況下,在(a)中,在給定母體分區中提供可釋放地連接至第一粒子的第一條形碼序列。在一些情況下,在(b)中,在給定父體分區中提供可釋放地連接至第二粒子的第二條形碼序列。
本公開的額外方面提供一種用於表徵樣品核酸的方法。在一些情況下,方法包括:(a)從受試者獲得生物樣品,所述生物樣品包括無細胞樣品核酸;(b)在微滴中,將條形碼序列連接至無細胞樣品核酸的片段或樣品核酸的諸多個部分的拷貝,以提供條形碼化樣品片段;(c)測定條形碼化樣品片段的核酸序列並且基於條形碼化樣品片段的核酸序列提供樣品核酸序列;(d)使用經過編程的計算機處理器來產生樣品核酸序列與參考核酸序列的比較,所述參考核酸序列具有大於10千鹼基(kb)的長度和至少99%的準確度;以及(e)使用所述比較來識別樣品核酸序列中的一種或多種基因變異,由此將樣品核酸與疾病相關聯。在一些情況下,樣品核酸序列中的一種或多種基因變異選自結構變異和單核苷酸多態性(SNP)。在一些情況下,樣品核酸序列的一種或多種基因變異為選自拷貝數變異、插入、缺失、反轉錄轉座子、易位、倒位、重排、重複擴增以及重複的結構變異。在一些情況下,在(c)中,以至少99%的準確度提供樣品核酸序列。在一些情況下,在(b)中,在微滴中提供可釋放地連接至粒子的條形碼序列,並且其中(b)進一步包括在連接條形碼序列之前使條形碼序列從粒子釋放至微滴中。在一些情況下,在(b)中,提供條形碼序列作為可釋放地連接至的粒子的引物序列的一部分,其中引物序列還包括隨機N-mer序列,並且其中(b)進一步包括在連接條形碼序列之前使引物序列從粒子釋放至微滴中。在一些情況下,在(b)中,在擴增反應中使用引物將條形碼序列連接至無細胞樣品核酸的片段或無細胞樣品核酸的諸多個部分的拷貝。
在一些情況下,用於表徵樣品核酸的方法進一步包括:(i)在額外微滴中,將額外條形碼序列連接至參考核酸的片段或參考核酸的諸多個部分的拷貝以提供條形碼化參考片段;以及(ii)測定條形碼化參考片段的核酸序列並且基於條形碼化參考片段的核酸序列測定參考核酸序列。在一些情況下,測定參考核酸序列包括組裝條形碼化參考片段的核酸序列。在一些情況下,用於表徵樣品核酸的方法進一步包括在額外微滴中提供可釋放地連接至粒子的額外條形碼序列以及在連接額外條形碼序列之前使額外條形碼序列從粒子釋放至額外分區中。在一些情況下,用於表徵樣品核酸的方法進一步包括提供額外條形碼序列作為可釋放地連接至粒子的引物序列的一部分,其中引物序列還包括隨機N-mer序列,並且在連接額外條形碼序列之前使引物從粒子釋放至額外微滴中。在一些情況下,用於表徵樣品核酸的方法進一步包括在擴增反應中使用引物將額外條形碼序列連接至參考核酸的片段或參考核酸的諸多個部分的拷貝。在一些情況下,用於表徵樣品核酸的方法進一步包括測定參考核酸序列中的一種或多種基因變異。
在一些情況下,參考核酸序列中的一種或多種基因變異選自結構變異和單核苷酸多態性(SNP)。在一些情況下,參考核酸序列中的一種或多種基因變異為選自拷貝數變異、插入、缺失、反轉錄轉座子、易位、倒位、重排、重複擴增以及複製的結構變異。在一些情況下,參考核酸包括生殖系核酸序列。在一些情況下,參考核酸包括癌症核酸序列。在一些情況下,樣品核酸序列具有大於10kb的長度。在一些情況下,參考核酸源於指示疾病狀態不存在的基因組。在一些情況下,參考核酸源於指示疾病狀態的基因組。在一些情況下,疾病狀態包括癌症。在一些情況下,疾病狀態包括非整倍性。在一些情況下,無細胞樣品核酸包括腫瘤核酸。在一些情況下,腫瘤核酸包括循環腫瘤核酸。
由以下詳細描述本公開的額外方面和優點對本領域技術人員來說將變得輕易顯而易見,其中僅示出和描述了本公開的說明性實施方案。如將認識到的,本公開能夠實現其他和不同實施方案,並且在各個明顯的方面其若干細節能夠進行修改,所有這些都不脫離本公開。因此,圖式和描述將被視為在本質上是說明性的,而不是限制性的。
以引用的方式併入
本說明書中提到的所有出版物、專利以及專利申請以全文引用的方式併入本文中,其程度如同每個單個出版物、專利或專利申請被具體地和單個地指示以引用的方式併入一般。
附圖簡述
本發明的新穎特徵在所附權利要求書中被特別闡述。通過參考以下使用本發明原理闡述說明性實施方案的詳細描述以及附圖將獲得對本發明的特徵和優點的更好的理解,在附圖中:
圖1提供使用常規方法與本文所描述的示例性方法和系統來識別和分析定相變體的示意性說明。
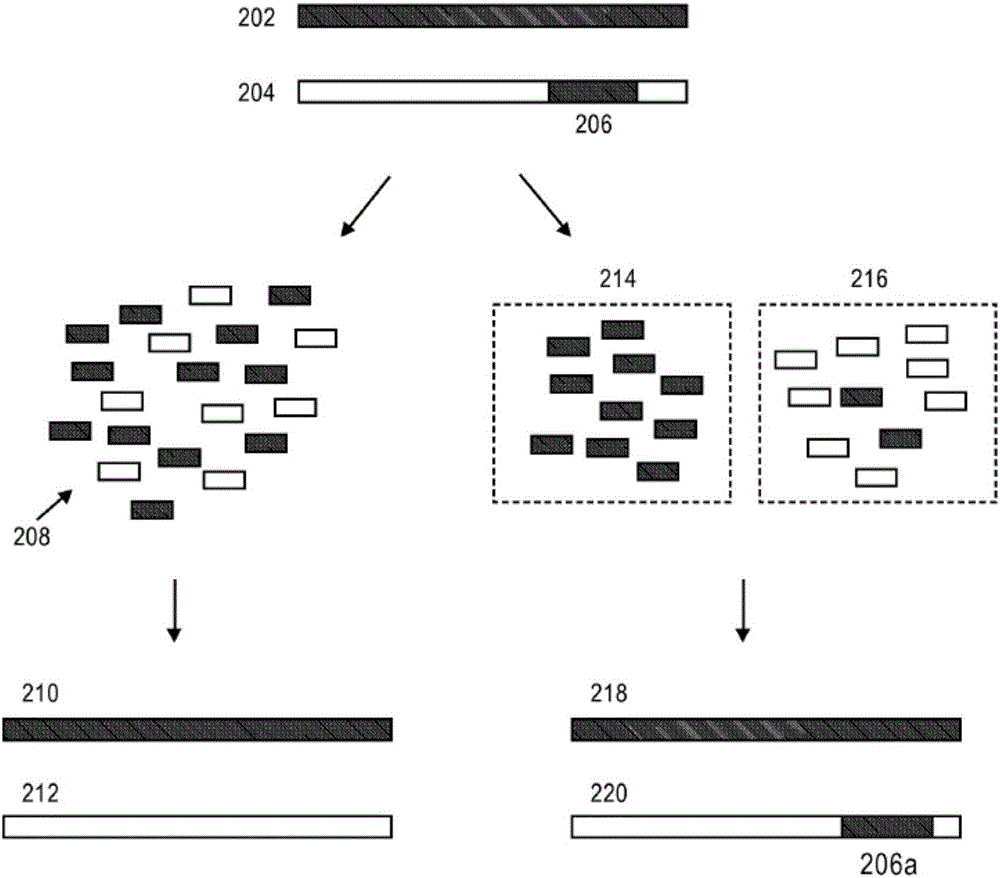
圖2提供使用常規方法與本文所描述的示例性方法和系統來識別和分析結構變異的示意性說明。
圖3說明使用本文所公開的方法和組合物進行分析來檢測拷貝數或單倍型的示例性工作流程。
圖4提供將核酸樣品與珠粒組合併且將核酸和珠粒分配至離散微滴中的示例性方法的示意性說明。
圖5提供條形碼化和擴增染色體核酸片段的示例性方法的示意性說明。
圖6提供條形碼化染色體核酸片段在將序列數據歸屬於單個染色體中的示例性用途的示意性說明。
圖7提供定相測序方法的實例的示意性說明。
圖8提供健康患者(上部圖)以及單倍型拷貝數增加(中部圖)或單倍型拷貝數減少(下部圖)的癌症患者的基因組的示例性子集的示意性說明。
圖9A-B提供:(a)示出腫瘤DNA的相對貢獻的示意性說明,以及(b)通過普通測序方法檢測此類拷貝增加和減少的圖示。
圖10提供使用單一變體位置(左側圖)和組合變體位置(右側圖)檢測拷貝增加和減少的實例的示意性說明。
圖11提供所描述的方法和系統用於識別拷貝數增加和減少的潛力的示意性說明。
圖12說明基於使用本文所描述的方法和組合物對染色體數目和拷貝數變異的測定來進行非整倍性測試的示例性工作流程。
圖13A-B說明用於識別基因樣品中的諸如易位和基因融合等結構變異的方法的示例性概括。
圖14說明基於使用本文所描述的方法和組合物對拷貝數變異的測定來進行癌症診斷測試的示例性工作流程。
圖15提供NCI-H2228癌症細胞系的EML-4-ALK結構變異的示意性說明。
圖16A和16B使用本文所描述的系統提供條形碼映射數據,以便識別與陰性對照細胞系(圖16B)相比在癌細胞系(圖16A)中存在圖15中所示的EML-4-ALK變體結構。
圖17示意性描述如本文所描述分析父體核酸序列的示例性工作流程。
圖18示意性描繪如本文所描述分析母體核酸序列的示例性工作流程。
圖19示意性描繪如本文所描述分析胎兒核酸序列的示例性工作流程。
圖20示意性描繪如本文所描述分析參考核酸序列的示例性工作流程。
圖21示意性描繪如本文所描述分析樣品核酸序列的示例性工作流程。
圖22示意性描繪示例性計算機控制系統。
詳細描述
雖然本文中已示出和描述了本發明的各個實施方案,但對本領域技術人員來說將顯而易見的是此類實施方案僅僅是通過舉例而提供。本領域技術人員會想到許多變化、改變以及替換,而不會脫離本發明。應了解,可採用對本文中所描述的本發明實施方案的各種替代方案。
如本文中所用,術語「生物體」通常是指連續活系統。生物體的非限制性實例包括動物(例如人、其他類型的哺乳動物、鳥類、爬行動物、昆蟲、本文中別處所描述的其他示例性類型的動物)、植物、真菌以及細菌。
如本文中所用,術語「重疊群」通常是指給定長度的連續核酸序列。連續序列可源於單個序列讀段,包括短讀段或長讀段序列讀段,或來自序列讀段的組裝物,所述序列讀段是比對過的並且基於讀段內的重疊序列被組裝或基於其他已知連接數據(例如如本文中別處所描述用共同條形碼標記)被定義為在片段內相連接。這些重疊序列讀段可同樣包括短讀段,例如小於500個鹼基,例如在一些情況下,約100至500個鹼基,並且在一些情況下,100至250個鹼基,或基於更長的序列讀段,例如大於500個鹼基、1000個鹼基或甚至大於10,000個鹼基。
I.概括
本公開提供適用於在基因材料的表徵方面提供顯著進步的方法和系統。在一些情況下,所述方法和系統可適用於提供使用通常可獲得的技術非常難或甚至不可能實現的基因表徵,包括例如單倍型定相、識別結構變異(例如缺失、重複、拷貝數變體、插入、倒位、反轉錄轉座子、易位、LTR、STR)以及多種其他適用表徵。
一般來說,本文所描述的方法和系統通過提供對長單個核酸分子的測序來完成以上目標,所述測序允許識別和使用長範圍變體信息,例如將變異與不同序列區段相聯繫,包括含有其他變異的序列區段,其在起源序列中隔開例如長於由短讀段測序技術所提供的顯著距離。然而,這些方法和系統實現這些目標,具有短讀段測序技術的極低的並且遠低於報告過的長讀段長度測序技術(例如單分子測序,諸如SMRT測序和納米孔測序技術)的測序誤差率的優點。
一般來說,本文所描述的方法和系統將長核酸分子區段化成更小片段,所述更小片段是使用高通量、更高確度短讀段測序技術可測序的,但此類區段化是以允許源於更小片段的序列信息被歸屬於更長的單個起源核酸分子的方式進行。通過將序列讀段歸屬於更長的起源核酸分子,可獲得關於所述更長核酸序列的顯著表徵信息,這是單獨由短序列讀段通常不能獲得的。如所提到的,此類表徵信息可包括單倍型定相、識別結構變異以及識別拷貝數變異。
本文所描述的方法和系統的優點是關於大量一般實例來描述。在第一實例中,使用本文所描述的方法和系統識別和表徵定相序列變體。圖1示意性說明了定相變體識別(phased variant calling)的挑戰以及通過本文所描述的方法提供的解決方案。如圖所示,圖I中的核酸102和104代表不同染色體(例如母體和父體遺傳染色體)的相同區域的兩個單倍體序列。各序列在表徵各單倍體序列的不同等位基因處包括一系列變體,例如核酸102上的變體106-114,以及核酸104上的變體116-122。由於其序列讀段非常短,所以大多數測序技術不能提供單個變體相對於同一單倍體序列上的其他變體的環境。另外,因為它們依賴於不分離單個分子組分(例如各單倍體序列)的樣品製備技術,所以不能識別各種變體的定相,例如變體所源於的單倍體序列。因此,這些短讀段技術不能將這些變體解析至其起源分子。圖IIa和IIIa示意性說明了使用此方法的困難。簡要地說,對圖IIa中所示的從兩個單倍體序列匯集的片段進行測序,產生大量短序列讀段124,並且組裝所得序列126(圖IIIa中所示)。如圖所示,因為沒有圖IIa中的更短序列讀段中的任一者的相對定相環境,所以在組裝過程中將不能解析兩個不同單倍體序列之間的變體。因此,圖IIIa中所示的所得組裝物產生單一共有序列組裝物126,包括所有變體106-122。
相比之下,並且如圖1的圖IIb中所示,如使用上文所描述的方法,本文所描述的方法和系統將更長核酸102和104分解或區段化成更短的可測序片段,但在那些片段存在下保留將其歸屬於其起源分子環境的能力。圖IIb中對此進行了示意性說明,其中將不同片段根據其起源分子環境進行分組或「區室化(compartmentalize)」。在本公開的上下文中,此分組可通過將片段物理分配至保留分子環境的諸多個組中以及標記那些片段以隨後能夠闡明環境中的一者或兩者來完成。
此分組被示意性說明為在分別代表來自核酸102和104的短序列讀段的群組128和130之間分派較短序列讀段。因為在整個測序過程中起源序列環境被保留,所以可將所述環境用於解析原始分子環境,例如分別定相序列102和104之間的各種變體106-114和116-122。
在另一示例性有利應用中,所述方法和系統適用於表徵使用短讀段序列技術通常不可識別或至少難以識別的結構變體。
圖2中參考簡單易位事件對此進行了示意性說明。如圖所示,基因組樣品可包括核酸,所述核酸包括易位事件,例如基因元件206從序列202至序列204的易位。此類易位可為多種不同易位類型中的任一種,包括例如不同染色體之間(無論是到相同的還是不同的區域)、同一染色體的不同區域之間的易位。
再次,如在上文的圖1中所說明的實例中,常規測序是通過將圖I中的序列202和204分解成小片段並且如圖IIa中所示從那些片段產生短序列讀段208而開始。因為這些序列片段208為相對短的,所以在組裝過程期間易位序列206的環境(即起源於相同或不同序列上的變體位置時)容易丟失。此外,由於其短讀段長度,經常預測在使用參考序列時序列組裝幾乎根據定義就不會反映結構變異。因此,將不變地無視易位序列206的適當位置而組裝短序列讀段208,並且實際上將如圖IIIa中所示組裝非變體序列210和212。
相比之下,使用本文所描述的方法和系統,為源於序列202和204的短序列讀段提供區室化,在圖IIb中被示出為群組214和216,其保留更小序列片段的原始分子分組,從而允許其組裝為圖IIIb中所示的序列218和220,從而允許歸屬回到起源序列202和204,並且識別易位變異,例如如圖IIIb中所說明的恰當序列組裝物218和220中的易位序列區段206a。
如上文所提到,本文所描述的方法和系統提供更長核酸的短序列讀段的單個分子環境。如本文中所用,單個分子環境是指特定序列讀段以外的序列環境,例如與鄰近或接近序列相關,所述鄰近或接近序列不包括在序列讀段本身內,並且因此,通常將使得它們不會被整個或部分地包括在短序列讀段中,例如約150個鹼基或對於成對讀段來說約300個鹼基的讀段。在一些方面,所述方法和系統提供短序列讀段的長範圍序列環境。此類長範圍環境包括給定序列讀段與彼此距離長於1千鹼基(kb)、長於5kb、長於10kb、長於15kb、長於20kb、長於30kb、長於40kb、長於50kb、長於60kb、長於70kb、長於80kb、長於90kb或甚至長於100kb之內或更長的序列讀段的關係或聯繫。通過提供更長範圍單個分子環境,本文所描述的方法和系統還提供長得多的推測分子環境。序列環境如本文所描述可包括更低解析環境,例如由將短序列讀段映射至單個更長分子或相連分子的重疊群;以及更高解析序列環境,例如由更長單個分子中例如具有單個分子的連續的所測定序列的較大部分的長範圍測序,其中此類所測定序列長於1kb、長於5kb、長於10kb、長於15kb、長於20kb、長於30kb、長於40kb、長於50kb、長於60kb、長於70kb、長於80kb、長於90kb或甚至長於100kb。如在序列環境的情況下,將短序列歸屬於更長核酸(例如均為單個長核酸分子或相連核酸分子的集合或重疊群)可包括將短序列映射至更長核酸區段以提供高水平序列環境,以及由短序列通過這些更長核酸提供組裝序列。
此外,雖然可利用與長單個分子相關的長範圍序列環境,但具有此類長範圍序列環境還允許推測基至更長範圍的序列環境。舉一個例子,通過提供上文所描述的長範圍分子環境,可識別長序列之中來自不同起源分子的重疊變體部分,例如定相變體、易位序列等,從而允許得到那些分子之間的推測聯繫。此類推測聯繫或分子環境在本文中被稱為「推測重疊群」。在一些情況下,當在定相序列的背景下論述時,推測重疊群通常可代表定相序列,例如在憑藉重疊定相變體的情況下,可推測長度大體上大於單個起源分子的定相重疊群。這些定相重疊群在本文中被稱為「相位區塊」。
通過以更長單分子讀段開始,可得到比使用短讀段測序技術或其他定相測序方法以其他方式將可達到的更長的推測重疊群或相位區塊。參見例如已出版的美國專利公布號2013/0157870,該專利公布的全部公開內容以全文引用的方式併入本文中。特定而言,使用本文所描述的方法和系統,可獲得N50(重疊群或相位區塊長度,對於所述重疊群或相位區塊長度,具有所述長度或更長的所有相位區塊或重疊群的集合含有所有重疊群或相位區塊的長度總和的至少一半,並且對於所述重疊群或相位區塊長度,具有所述長度或更短的所有重疊群或相位區塊的集合也含有所有重疊群或相位區塊的長度總和的至少一半)、眾數、平均值或中值為至少約10千鹼基(kb)、至少約20kb、至少約50kb的推測重疊群或相位區塊長度。在一些方面,推測重疊群或相位區塊長度的N50、眾數、平均值或中值為至少約100kb、至少約150kb、至少約200kb,並且在一些情況下為至少約250kb、至少約300kb、至少約350kb、至少約400kb,並且在一些情況下達到至少約500kb、至少約750kb、至少約1Mb、至少約1.75Mb、至少約2.5Mb或更大。在其他情況下,可獲得至少或超過20kb、40kb、50kb、100kb、200kb、300kb、400kb、500kb、750kb、1兆鹼基(Mb)、1.75Mb、2Mb或2.5Mb的最大推測重疊群或相位區塊長度。在其他情況下,推測重疊群或相位區塊長度可為至少約20kb、至少約40kb、至少約50kb、至少約100kb、至少約200kb,並且在一些情況下為至少約500kb、至少約750kb、至少約1Mb,並且在一些情況下為至少約1.75Mb、至少約2.5Mb或更長。
在一個方面,本文所描述的方法和系統提供樣品核酸或其片段區室化、沉積或分配至離散區室或分區(本文中可互換地稱為分區)中,其中各分區保持其自己的內容物與其他分區的內容物隔開。可事先、隨後或同時將獨特的標識(例如條形碼)遞送至容納被區室化或分配的樣品核酸的分區,以允許隨後將特徵(例如核酸序列信息)歸屬於特定區室內所包括的樣品核酸,並且特別是歸屬於可能最初沉積至分區中的相對長的連續樣品核酸區段。
可對樣品核酸進行分配使得核酸以相對長的連續核酸分子片段或區段存在於分區中。這些片段可代表所要分析的全部樣品核酸的許多重疊片段,例如整個染色體、外顯子組或其他大基因組片段。這些樣品核酸可包括全基因組、單個染色體、外顯子組、擴增子或所關注的多種不同核酸中的任一種。在一些情況下,樣品核酸的這些片段可長於100個鹼基、長於500個鹼基、長於1kb、長於5kb、長於10kb、長於15kb、長於20kb、長於30kb、長於40kb、長於50kb、長於60kb、長於70kb、長於80kb、長於90kb或甚至長於100kb,這允許上文所描述的更長範圍的分子環境。
還可以使得給定分區包括起始樣品核酸的兩個重疊片段的概率極低的水平對樣品核酸進行分配。這可通過在分配過程期間以低輸入量和/或濃度提供樣品核酸來完成。因此,在一些情況下,給定分區可包括起始樣品核酸的許多長但不重疊的片段。然後將不同分區中的樣品核酸與獨特的標識相關聯,其中對於任何給定分區,其中所含的核酸具有相同的獨特標識,但其中不同分區可包括不同的獨特標識。另外,因為分配將樣品組分分派至極小體積的分區或微滴中,應了解為實現如上文所闡述的分派,不需要如在更高體積方法中(例如在管或多孔板的孔中)可能會要求的對樣品進行實質性稀釋。此外,因為本文所描述的系統採用如此高水平的條形碼多樣性,可如上文所提供將多樣的條形碼分派在更高數目的基因組當量之中。特定而言,先前所描述的多孔板方法(參見例如美國專利公布號2013/0079231和2013/0157870,這些美國專利公布的全部公開內容以全文引用的方式併入本文中)僅可使用一百至數百個不同的條形碼序列來操作,並且採用其樣品的有限稀釋過程以便能夠將條形碼歸屬於不同的細胞/核酸。因此,它們通常使用遠少於100個細胞來操作,這將會提供大約1:10並且當然遠高於1:100的基因組:(條形碼類型)比率。另一方面,本文所描述的系統由於高水平的條形碼多樣性(例如超過10,000、100,000、500,000(等)種多樣的條形碼類型)而可在大約1:50或更小、1:100或更小、1:1000或更小或甚至更小比率的基因組:(條形碼型)比率下操作,同時還允許在仍提供提高很多的每一基因組條形碼多樣性的同時加載更高數目的基因組(例如大約每次分析大於100個基因組、每次分析大於500個基因組、每次分析1000個基因組或甚至更多)。
經常,在分配之前將樣品與可釋放地連接至珠粒的寡核苷酸標籤的集合組合。寡核苷酸可包含至少第一和第二區域。第一區域可為條形碼區域,其在給定分區內的寡核苷酸之間可大體上為相同的條形碼序列,但在不同分區之間可能並且在大多數情況下為不同的條形碼序列。第二區域可為N-mer(例如隨機N-mer或被設計成靶向特定序列的N-mer),其可用於引導分區內的樣品內的核酸。在一些情況下,在N-mer被設計成靶向特定序列的情況下,其可被設計成靶向特定染色體(例如染色體1、13、18或21)或染色體區域,例如外顯子組或其他靶向區域。在一些情況下,N-mer可被設計成靶向特定基因或基因區域,諸如與疾病或病症(例如癌症)相關的基因或區域。在分區內,可使用第二N-mer進行擴增反應以在沿核酸長度的不同位置引導核酸樣品。由於擴增,各分區可含有核酸的擴增產物,所述核酸的擴增產物連接至同一或近乎同一的條形碼,並且在各分區中可呈現核酸的重疊的更小片段。條形碼可充當標記物,所述標記物表示核酸的集合起源於同一分區,並且因此可能也起源於核酸的同一鏈。在擴增之後,可匯集核酸,使用測序算法進行測序和比對。因為更短序列讀段可憑藉其相關條形碼序列進行比對並且被歸屬於樣品核酸的單一長片段,所以可將所述序列上所有所識別的變體歸屬於單一起源片段和單一起源染色體。此外,通過比對多個長片段上的多個共定位變體,可進一步表徵所述染色體貢獻。因此,然後可得出關於特定基因變體定相的結論。此類信息可適用於識別單倍型,所述單倍型通常為位於同一核酸鏈上或不同核酸鏈上的指定基因變體集合。拷貝數變異也可以此方式進行識別。
所描述的方法和系統提供優於當前核酸測序技術和其相關樣品製備方法的顯著優點。因為對生物樣品(例如血液、細胞或組織樣品)進行整體處理來從細胞整體提取基因材料,並且將其轉化成被配置成特定用於給定測序技術的測序文庫,所以通過對基因組DNA進行測序通常不可獲得單倍型定相和拷貝數變異數據。由於此整體樣品處理方法,測序數據通常提供非定相基因型,以此方式不可能確定基因信息是存在於同一染色體還是不同染色體上。
除了不能將基因特徵歸屬於特定染色體,此類整體樣品製備和測序方法還傾向於主要識別和表徵樣品中的多數組分,並且未被設計成識別和表徵少數組分,例如由一個染色體或由一個或數個細胞所貢獻的基因材料或構成所提取樣品中的全部DNA的較小百分比的在血流中循環的片段化腫瘤細胞DNA分子。所描述的方法和系統還提供檢測存在於更大樣品中的較小群體的顯著優點。因此,它們可適用於評估樣品中的拷貝數變異,因為經常僅一小部分的臨床樣品含有具有拷貝數變異的組織。舉例來說,如果樣品為來自孕婦的血液樣品,那麼僅一小部分的樣品會含有循環無細胞胎兒DNA。
本文所公開的條形碼化技術的使用賦予所述技術提供給定基因標記物集合的單個分子環境的能力,即將給定基因標記物集合(與單一標記物不同)歸屬於單個樣品核酸分子,並且通過變體協調性組裝來在多個樣品核酸分子之中提供更寬或甚至更長範圍的推測單個分子環境;和/或歸屬於特定染色體的能力。這些基因標記物可包括特定基因座,例如變體,諸如SNP;或者它們可包括短序列。此外,條形碼化的使用賦予以下額外優點:促成區分從樣品提取例如用於檢測和表徵血流中的循環腫瘤DNA的全部核酸群體中的少數組分與多數組分以及減少或消除任何擴增期間的擴增偏向的能力。另外,以微流體模式實現賦予以極小樣品體積和低輸入量的DNA工作的能力,以及快速處理大量樣品分區(例如微滴)以促進基因組範圍標記的能力。
如先前所描述,本文所描述的方法和系統的優點在於它們可通過使用廣泛可用的短讀段測序技術來實現結果。此類技術具有以下優點:可輕易獲得並且在研究界內分布範圍很廣,並且具有充分表徵和高度有效的方案和試劑系統。這些短讀段測序技術包括可從例如Illumina,Inc.(例如GXII、NextSeq、MiSeq、HiSeq、X10)、Thermo-Fisher的Ion Torrent分公司(例如Ion Proton和Ion PGM)獲得的那些、焦磷酸測序法以及其他技術。
特別有利的是,本文所描述的方法和系統利用這些短讀段測序技術並且這樣做具有相關低誤差率。特定而言,如上文所描述,本文所描述的方法和系統實現單個分子讀段長度或環境,但具有單個測序讀段,從而排除mate pair延伸,其短於1000bp、短於500bp、短於300bp、短於200bp、短於150bp或甚至更短;並且對於此類單個分子讀段長度來說,具有低於5%、低於1%、低於0.5%、低於0.1%、低於0.05%、低於0.01%、低於0.005%或甚至低於0.001%的測序誤差率。
II.工作流程概括
在一個示例性方面,本公開中所描述的方法和系統使得單個樣品(例如核酸)沉積或分配至離散分區中,其中各分區維持其自身的內容物與其他分區的內容物分離。如本文中所用,分區是指器皿或容器,其可包括多種不同形式,例如孔、管、微孔或納米孔、通孔等。然而,在一些方面,分區可在流體流內流動。這些容器可包含例如具有包圍內部流體中心或核心的外部屏障的微膠囊或微囊泡,或其可為能夠夾帶和/或保留基質內的材料的多孔基質。然而,在一些方面,這些分區可包含非水性連續相(例如油相)內的水性流體的微滴。多種不同的容器描述於例如2013年8月13日提交的美國專利申請號13/966,150中。同樣地,用於形成非水性或油性連續相中的穩定微滴的乳液系統詳細描述於例如美國專利公布號2010/0105112中,所述專利公布的全部公開內容以全文引用的方式併入本文中。在某些情況下,微流體通道網絡可適合用於產生如本文所描述的分區。此類微流體裝置的實例包括詳細描述於2014年4月10日提交的美國臨時專利申請號61/977,804中的那些,所述臨時專利申請的全部公開內容出於所有目的以全文引用的方式併入本文中。在分配單個細胞時還可採用替代機制,包括多孔膜,細胞的水性混合物穿過所述多孔膜被擠壓至非水性流體中。此類系統通常可自例如Nanomi,Inc.獲得。
在乳液中的液滴的情況下,將樣品材料(例如核酸)分配至離散分區中通常可通過以下方式完成:使水性的含有樣品的流流至接頭中,也使分配流體(例如氟化油)的非水性流流至所述接頭中,使得在流動的流分配流體內形成水性微滴,其中此類微滴包括樣品材料。如下所述,分區(例如微滴)還可包括共分配的條形碼寡核苷酸。可通過控制系統的各種不同參數來調節任何特定分區內的樣品材料的相對量,所述各種不同參數包括例如水性流中的樣品濃度、水性流和/或非水性流的流速等。本文所描述的分區的特徵常常為具有極小的體積。舉例來說,在基於微滴的分區的情況下,微滴可具有小於1000皮升(pL)、小於900pL、小於800pL、小於700pL、小於600pL、小於500pL、小於400pL、小於300pL、小於200pL、小於100pL、小於50pL、小於20pL、小於10pL或甚至小於1pL的總體積。在與珠粒共分配的情況下,應了解,分區內的樣品流體體積可為上文所描述的體積的小於90%、小於80%、小於70%、小於60%、小於50%、小於40%、小於30%、小於20%或甚至為上文所描述的體積的小於10%。在一些情況下,在與極小量的起始試劑(例如輸入核酸)進行反應時,使用低反應體積分區可為有利的。用於在低輸入核酸情況下分析樣品的方法和系統提供於2014年6月26日提交的美國臨時專利申請號62/017,580中,該臨時專利申請的全部公開內容以全文引用的方式併入本文中。
一旦將樣品引入其各自的分區中,根據本文所描述的方法和系統,分區內的樣品核酸通常具備獨特的標識,使得在表徵那些核酸之後,可將其歸因為源於其各自的起源。因此,可將樣品核酸與獨特的標識(例如條形碼序列)共分配。在一些方面,以包含可連接至那些樣品的核酸條形碼序列的寡核苷酸的形式來提供獨特的標識。對寡核苷酸進行分配,使得在給定分區中的寡核苷酸之間,其中所含的核酸條形碼序列相同,但在不同分區之間,寡核苷酸可具有不同的條形碼序列。在一些方面,僅一個核酸條形碼序列可與給定分區相關聯,不過在一些情況下,可存在兩個或更多個不同的條形碼序列。
核酸條形碼序列可在寡核苷酸的序列內包括6至約20個或更多個核苷酸。這些核苷酸可為完全連續的,即呈單段相鄰核苷酸的形式,或者它們可被分隔至由一個或多個核苷酸隔開的兩個或更多個單獨的子序列中。在一些情況下,隔開的子序列的長度可為約4至約16個核苷酸。
共分配的寡核苷酸還可包含適用於處理共分配的核酸的其他功能序列。這些序列包括例如靶向型或隨機/通用型擴增引物序列,其用於擴增分區內的單個細胞的基因組DNA,同時連接相關條形碼序列、測序引物、雜交或探測序列,例如用於識別序列的存在或用於向下拉動條形碼化核酸;或許多其他潛在功能序列中的任一種。再次,寡核苷酸和相關條形碼以及其他功能序列連同樣品材料的共分配描述於例如2014年2月7日提交的美國臨時專利申請號61/940,318和2014年5月9日提交的美國臨時專利申請號61/991,018以及2014年6月26日提交的美國專利申請號14/316,383以及2014年2月7日提交的美國專利申請號14/175,935中,這些專利申請的全部公開內容以全文引用的方式併入本文中。
簡要地說,在一種示例性方法中,提供珠粒,所述珠粒各自可包括大量上文所描述的可釋放地連接至珠粒的寡核苷酸,其中連接至特定珠粒的所有寡核苷酸可包括相同的核酸條形碼序列,但其中在所用的珠粒群體中可呈現大量多樣的條形碼序列。在一些情況下,珠粒群體可提供多樣的條形碼序列文庫,其可包括至少1000個不同的條形碼序列、至少10,000個不同的條形碼序列、至少100,000個不同的條形碼序列或在一些情況下,至少1,000,000個不同的條形碼序列。另外,各珠粒可具備所連接的大量寡核苷酸分子。特定而言,單個珠粒上包括條形碼序列的寡核苷酸分子的數目可為至少約10,000個寡核苷酸、至少100,000個寡核苷酸分子、至少1,000,000個寡核苷酸分子、至少100,000,000個寡核苷酸分子,且在一些情況下為至少十億個寡核苷酸分子。
在對珠粒施加特定刺激後,寡核苷酸可從珠粒釋放。在一些情況下,刺激可為光刺激,例如通過可釋放寡核苷酸的光不穩定性鍵的裂解。在一些情況下,可使用熱刺激,其中珠粒環境的溫度升高可能會導致鍵的裂解或寡核苷酸從珠粒的其他釋放。在一些情況下,可使用化學刺激,從而裂解寡核苷酸與珠粒的鍵,或以其他方式可使得寡核苷酸從珠粒釋放。
根據本文所描述的方法和系統,可將包括連接的寡核苷酸的珠粒與單個樣品共分配,使得單個分區內含有單一珠粒和單一樣品。在一些情況下,在需要單珠粒分區的情況下,可控制流體的相對流速,使得所述分區平均每個分區含有少於一個珠粒,以確保被佔用的那些分區主要是單一佔用的。同樣地,可能希望控制流速以使得更高百分比的分區被佔用,從而例如僅允許較小百分比的未佔用分區。在一些方面,控制流量和通道結構,以確保所需數目的單一佔用分區、低於某一水平的未佔用分區以及低於某一水平的多重佔用分區。
圖3說明用於條形碼化樣品核酸並且隨後測序諸如用於拷貝數變異或單倍型分析的示例性方法。首先,可從來源獲得包含核酸的樣品300,並且還獲得條形碼化珠粒的集合310。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將較低量的包含核酸的樣品305、條形碼化珠粒315以及(在一些情況下)其他試劑(例如還原劑)320組合併且進行分配。舉例來說,此類分配可涉及將組分引入微滴產生系統,諸如微流體裝置325。在微流體裝置325的輔助下,可形成油包水乳液330,其中所述乳液含有水性微滴,所述水性微滴含有樣品核酸305、還原劑320以及條形碼化珠粒315。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放335。隨機N-mer可然後引導樣品核酸的不同區域,從而在擴增之後產生樣品的擴增拷貝,其中將各拷貝用條形碼序列標記340。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。隨後,將乳液破壞345,並且可經由例如擴增方法350(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可進行測序355,並且應用算法來解釋測序數據360。測序算法通常能夠例如對條形碼進行分析以比對測序讀段和/或識別特定序列讀段所屬的樣品。
如上文所提到,雖然單一珠粒佔用可能為所需的,但是應了解可能經常存在多重佔用分區或未佔用分區。圖4中示意性說明了用於對樣品和包含條形碼寡核苷酸的珠粒進行共分配的微流體通道結構的實例。如圖所示,以在通道接頭412處流體連通的形式提供通道區段402、404、406、408以及410。使包含單個樣品414的水性流通過通道區段402流至通道接頭412。如本文別處所描述,可在分配過程之前將這些樣品懸浮於水性流體內。
同時,使包含攜帶條形碼的珠粒的水性流416通過通道區段404流至通道接頭412。將非水性分配流體從側通道406和408中的每一者引入通道接頭412中,並且使組合流流至出口通道410中。在通道接頭412內,將來自通道區段402和404的兩條組合水性流組合在一起,並且分配至微滴418中,所述微滴包括共分配的樣品414和珠粒416。如前面提到的,通過控制在通道接頭412處組合的各個流體的流動特徵,以及控制通道接頭的幾何結構,可優化組合和分配以實現珠粒、樣品或兩者在所產生的分區418內的所需佔用水平。
如應了解,可將許多其他試劑與樣品和珠粒一起共分配,包括例如化學刺激物;核酸延伸、轉錄和/或擴增試劑,諸如聚合酶、反轉錄酶、三磷酸核苷或NTP類似物、引物序列和額外輔因子(諸如用於此類反應中的二價金屬離子)、連接反應試劑(諸如連接酶和連接序列);染料、標籤或其他標記試劑。
一旦共分配,即可使用設置於珠粒上的寡核苷酸來對所分配的樣品進行條形碼化和擴增。在對樣品進行擴增和條形碼化時使用這些條形碼寡核苷酸的示例性方法詳細描述於2014年2月7日提交的美國專利申請號61/940,318和2014年5月9日提交的美國專利申請號61/991,018以及2014年6月26日提交的美國專利申請號14/316,383中,這些專利申請的全部公開內容以全文引用的方式併入本文中。簡要地說,在一個方面,寡核苷酸存在於與樣品共分配的珠粒上並且與樣品一起從其珠粒釋放至分區中。寡核苷酸可(連同條形碼序列一起)在其5』端包括引物序列。此引物序列可為意在隨機引導樣品的許多不同區域的隨機寡核苷酸序列或其可為以引導樣品的特定靶向區域的上遊為目標的特定引物序列。
一旦被釋放,寡核苷酸的引物部分即可與樣品的互補區域退火。也與樣品和珠粒共分配的延伸反應試劑(例如DNA聚合酶、三磷酸核苷、輔因子(例如Mg2+或Mn2+等))然後使用樣品作為模板來延伸引物序列,以產生與引物退火的模板鏈的互補片段,其中互補片段包括寡核苷酸和其相關條形碼序列。多個引物與樣品的不同部分的退火和延伸可產生樣品的重疊互補片段的大型匯集物,所述重疊互補片段各自具有其自己的指示其在其中形成的分區的條形碼序列。在一些情況下,這些互補片段本身可用作模板,所述模板由存在於分區中的寡核苷酸引導以產生互補序列的互補序列,其又包括條形碼序列。在一些情況下,此複製過程被配置為使得當第一互補序列重複時,其產生位於或靠近其末端的兩個互補序列,以允許形成髮夾結構或部分髮夾結構,從而降低所述分子成為產生其他重複拷貝的基礎的能力。圖5中示出了對此情況的一個實例的示意性說明。
如該圖所示,將包括條形碼序列的寡核苷酸與樣品核酸504一起共分配於例如乳液中的微滴502中。如本文別處所提到,如圖A中所示,寡核苷酸508可提供於與樣品核酸504共分配的珠粒506上,所述寡核苷酸可從珠粒506釋放。寡核苷酸508除一個或多個功能序列(例如序列510、514以及516)之外還包括條形碼序列512。舉例來說,寡核苷酸508被示出為包含條形碼序列512以及可充當給定測序系統的連接或固定序列的序列510,例如用於在Illumina Hiseq或Miseq系統的流動細胞中進行連接的P5序列。如圖所示,寡核苷酸還包括引物序列516,其可包括用於引導樣品核酸504的諸多個部分的複製的隨機或靶向型N-mer。寡核苷酸508內還包括序列514,其可提供測序引導區,諸如「讀段1」或R1引導區,所述引導區用於通過測序系統中的合成反應來引導聚合酶介導的模板定向測序。在一些情況下,條形碼序列512、固定序列510以及R1序列514對於連接至給定珠粒的所有寡核苷酸來說可為共同的。引物序列516可能因隨機N-mer引物而不同,或者在某些靶向應用中對於給定珠粒上的寡核苷酸來說可為共同的。
基於存在引物序列516,寡核苷酸能夠如圖B中所示引導樣品核酸,這允許使用也與珠粒506和樣品核酸504共分配的聚合酶和其他延伸試劑來延伸寡核苷酸508和508a。如圖C中所示,在對於隨機N-mer引物來說將與樣品核酸504的多個不同區域退火的寡核苷酸延伸之後;形成核酸的多個重疊互補序列或片段,例如片段518和520。雖然包括與樣品核酸的諸多個部分互補的序列部分,例如序列522和524,但是這些構建體在本文中通常被稱為包含樣品核酸504中具有連接的條形碼序列的片段。如應了解,如上文所描述的模板序列的複製部分在本文中經常被稱為所述模板序列的「片段」。然而,儘管如此,術語「片段」涵蓋起源核酸序列(例如模板或樣品核酸)的一部分的任何表示,包括通過提供模板序列的諸多個部分的其他機制(諸如給定序列分子的實際片段化,例如通過酶促、化學或機械片段化)所形成的那些。然而,在一些方面,模板或樣品核酸序列的片段可表示基礎序列的複製部分或其互補序列。
然後可例如通過序列分析對條形碼化核酸片段進行表徵,或可如在如圖D中所示的過程中將其進一步擴增。舉例來說,也從珠粒306釋放的額外寡核苷酸(例如寡核苷酸508b)可引導片段518和520。特定而言,再次,基於隨機N-mer引物516b存在於寡核苷酸508b中(這在一些情況下可不同於給定分區中的其他隨機N-mer,例如引物序列516),寡核苷酸與片段518退火,並且延伸以形成片段518中包括序列528的至少一部分的互補序列526,其包含樣品核酸序列的一部分的重複。寡核苷酸508b繼續延伸直到它已通過片段518的寡核苷酸部分508複製。如本文別處所提到,並且如圖D中所說明,寡核苷酸可被配置成提示通過聚合酶進行的複製在所需點停止,例如在通過寡核苷酸508的包括在片段518內的序列516和514複製之後停止。如本文所描述,這可通過不同方法來實現,包括例如併入不能由所用的聚合酶處理的不同核苷酸和/或核苷酸類似物。舉例來說,這可包括在序列區域512內納入含尿嘧啶的核苷酸來防止非尿嘧啶耐受型聚合酶使所述區域的複製停止。結果,形成片段526,其在一個末端包括全長寡核苷酸508b,包括條形碼序列512、連接序列510、R1引物區514以及隨機N-mer序列516b。在序列的另一個末端可包括第一寡核苷酸508的隨機N-mer的互補序列516』,以及整個或一部分的R1序列的互補序列以(以序列514』示出)。R1序列514和其互補序列514』然後能夠雜交在一起以形成部分髮夾結構528。如應了解,因為不同寡核苷酸之間的隨機N-mer不同,這些序列和其互補序列預期不會參與髮夾形成,例如序列516』(其為隨機N-mer 516的互補序列)預期不會與隨機N-mer序列516b互補。對於其他應用來說不會是這種情況,例如靶向型引物,其中在給定分區內寡核苷酸之間的N-mer將為共同的。
通過形成這些部分髮夾結構,允許從進一步的複製中去除樣品序列的第一級重複,從而例如防止拷貝的重複拷貝。部分髮夾結構還提供適用於所形成的片段(例如片段526)的後續處理的結構。
可然後如本文所描述從多個不同分區匯集所有片段以便在高通量測序儀上進行測序。因為各片段是關於其起源分區而被編碼,所以基於條形碼的存在可將所述片段的序列歸屬回到其起源。圖6中對這進行了示意性說明。如一個實例中所示,將起源於第一來源600(例如單個染色體、核酸鏈等)的核酸604和源於不同染色體602或核酸鏈的核酸606各自如上文所描述與其自己的條形碼寡核苷酸集合一起分配。
在各分區內,各核酸604和606然後被處理,以單獨地提供第一片段的重疊的第二片段集合,例如第二片段集合608和610。此處理還提供第二片段,其中來源於特定第一片段的第二片段中的每一者的條形碼序列是相同的。如圖所示,第二片段集合608的條形碼序列由「1」表示,而片段集合610的條形碼序列由「2」表示。可使用多樣的條形碼文庫來區別地條形碼化大量不同片段集合。然而,沒有必要用不同的條形碼序列來條形碼化來自不同第一片段的每一個第二片段集合。在一些情況下,可同時處理多個不同的第一片段以包括相同的條形碼序列。本文在別處詳細描述了多樣的條形碼文庫。
然後可匯集例如來自片段集合608和610的條形碼化片段,以便使用例如通過可從Illumina或Thermo Fisher,Inc.的Ion Torrent分公司獲得的合成技術獲得的序列進行測序。一旦經過測序,即可將序列讀段612至少部分基於所包括的條形碼並且在一些情況下部分基於其片段的序列而歸屬於其各自的片段集合,例如如聚集讀段614和616中所示。然後組裝被歸屬於各片段集合的序列讀段以提供各樣品片段的組裝序列,例如序列618和620,所述組裝序列又可被進一步歸屬回到其各自的原始染色體(600和602)。用於組裝基因組序列的方法和系統描述於例如2014年6月26日提交的美國臨時專利申請號62/017,589中,該臨時專利申請的全部公開內容以全文引用的方式併入本文中。在一些實例中,通過從頭組裝和/或基於參考序列的組裝(例如映射至參考序列)來組裝基因組序列。
III.將方法和系統應用於定相和拷貝數分析
在本文所描述的系統和方法的一個方面,將序列讀段歸屬於更長起源分子的能力用於測定關於序列的相位信息。在一個實例中,比較與顯示兩個或更多個特定基因變體序列(例如等位基因、基因標記物)的序列相關的條形碼以確定基因標記物的集合是否位於樣品中的同一染色體或不同染色體上。可使用此類定相信息來確定樣品中某些靶染色體或基因的相對拷貝數。所描述的方法和系統(symptom)的優點在於可使用多個位置、基因座、變體等來識別它們所起源於的單個染色體或核酸鏈以測定定相和拷貝數信息。經常,使用沿染色體的多個位置(例如大於2、3、4、5、6、7、8、9、10、20、30、40、50、100、500、1000、5000、10000、50000、100000或500000個)來測定本文所描述的定相、單倍型以及拷貝數變異信息。
舉例來說,如上文所提到,儘管利用可提供相對更短的序列讀段的測序技術,本文所描述的方法和系統憑藉上文所描述的分配和歸屬方面可適用於從單個核酸片段(例如單個核酸分子)提供有效長序列讀段。因為這些長序列讀段可被歸屬於單一起始片段或分子,所以序列中的變體位置可同樣被歸屬於單一分子,並且通過外推法歸屬於單一染色體。另外,可採用任何給定片段上的多個位置作為鄰近片段的比對特徵來提供可被推測為起源於同一染色體的比對序列。舉例來說,可對第一片段進行測序,並且憑藉上文所描述的歸屬方法和系統,可將存在於所述序列上的變體全部歸屬於單一染色體。然後可將共有被確定為僅存在於一個染色體上的多個這些變體的第二片段假定為源於同一染色體,並且因此與第一片段比對,以形成兩個片段的定相比對。重複此過程允許識別長範圍相位信息。可從已知參考物(例如HapMap)或從例如顯示以其他方式同一的序列區段上的不同變體的測序數據的集合獲得單一染色體上的變體的識別。
圖7提供示例性定相測序方法的示意性說明。如圖所示,可將起源核酸702(諸如染色體、染色體片段、外顯子組或其他大的單核酸分子)片段化成多個大片段704、706、708。起源核酸702可包括特定核酸分子(例如染色體)所特有的許多序列變體(A、B、C、D、E、F以及G)。根據本文所描述的方法,可將起源核酸片段化成多個大的重疊片段704、706以及708,其包括相關序列變體的子集。然後可如本文所描述將各片段分配,進一步片段化成子片段,並且條形碼化,以提供更大片段的多個重疊的條形碼化子片段,其中給定更大片段的子片段帶有相同的條形碼序列。舉例來說,與條形碼序列「1」和條形碼序列「2」相關的子片段分別顯示於分區710和712中。然後可匯集條形碼化的子片段,測序,並且組裝測序過的子片段以提供長片段序列714、716以及717。長片段序列714、716以及717中的一者或多者可包括多個變體。然後可基於序列714、716以及717的重疊定相變體信息進一步組裝長片段序列以提供定相序列718,從所述定相序列可確序定相位置。
一旦確定了定相位置,即可進一步以多種方式來探究信息。舉例來說,可利用評估某些病症的基因風險時對定相變體的了解,識別父體和母體特徵,識別非整倍性,或識別單倍型分析信息。
在本文所公開的系統和方法的一些方面,使用同時檢測兩種或更多種定相基因標記物來進行拷貝數變異分析以提高拷貝數計數的準確度。與在原初方法下僅基於多個基因座上和單倍型之間的計數讀段的變異相比,利用定相信息可增加信號的相對強度。另外,利用定相信息允許位置特異性偏向的正規化,從而大體上進一步增強信號。拷貝數變異(CNV)準確度可取決於眾多因素,包括測序長度、CNV長度、拷貝數目等)。本文所提供的方法和系統可以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%或99.999%的準確度測定CNV。在一些情況下,本文所提供的方法和系統以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%或0.000005%的誤差率測定CNV。類似地,本文所提供的方法和系統可以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%或99.999%的準確度檢測兩個或更多個基因變體的定相/單倍型信息。在一些情況下,本文所提供的方法和系統以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%或0.000005%的誤差率測定定相或單倍型信息。本公開還提供去除基因座特異性偏向的方法,其中基因座特異性變異減少了至少2倍、3倍、4倍、5倍、10倍、20倍、30倍、40倍、50倍、60倍、70倍、80倍、90倍、100倍、200倍、500倍、1000倍、5000倍或10000倍。可使用本文所提供的方法和系統來檢測拷貝數變異,諸如在拷貝數變化反映染色體數目或染色體的諸多個部分的變化的情況下。在一些情況下,可使用本文所提供的方法和系統來檢測存在於同一染色體上的基因的拷貝數變異。
圖8(上部圖)為說明健康患者的基因組的子集的示意圖。此患者具有在所示基因座處的雜合基因型和位於單獨染色體鏈上的兩個單獨的單倍型(1和2)805、810。患者的天然存在的變異(諸如SNP或缺失)以圓圈描繪。圖8還描繪癌症患者的基因組815。某些癌症與單倍型拷貝數增加相關。中部圖描繪單倍型2增加810。如圖8中的底部圖中所示顯示單倍型2減少820的底部圖中所示,癌症也可能與單倍型數目減少相關。常用測序技術不能準確確定此單倍型拷貝減少或增加。如圖9a中所示,這是部分歸因於以下事實:患者血液中的腫瘤貢獻的DNA 910僅為全部DNA中的一小部分,全部DNA中多數是由正常組織貢獻的DNA905。腫瘤DNA的此低濃度使得在一般測序技術下對拷貝數的檢測不精確,參見圖9b。難以檢測無拷貝變異920在平均深度D 935處的預期計數的峰與拷貝減少925(940)和拷貝增加930(945)的峰的差異。對於任何給定的單個標記物,在重複測試中拷貝數分析的結果的分布可以近似於泊松分布的方式公布於正確答案周圍,其中分布的寬度取決於分析中的隨機誤差的各種來源。因為對於給定樣品來說,拷貝數的變化可為樣品中相對小的部分,所以監測單一基因標記物時的寬的概率分布會掩蓋正確的結果。此困難是歸因於以下事實:如圖10(左側圖)中所示,一般測序技術一次僅關注單倍型的一個單一變體位置。使用此類技術,在代表拷貝減少1025、正常拷貝1020以及拷貝增加1030的峰之間可存在顯著重疊。本文所公開的技術允許檢測全部(或部分)單倍型、增加解析率以及改善對拷貝增加和減少的檢測,圖10(右側圖)。圖11中示意性示出此改善,其中一般檢測1100產生展開的重疊峰,而本文中的技術1110允許更精細的峰和對拷貝增加或減少的改善的解析。使用同時監測兩種或更多種定相基因標記物,特別是已知共定位於單一染色體上並且可能因此最有可能總是以同步化非隨機方式以更大或更小數目出現的標記物,具有使預期結果分布的寬度變窄並且同時提高計數準確度的作用。
除了在檢測和診斷癌症方面的優點,本文所提供的方法和系統還提供更準確和靈敏的檢測胎兒非整倍性的方法。
胎兒非整倍性為胎兒染色體數目失常。非整倍性通常引起顯著的物理和神經損傷。舉例來說,X染色體數目減少引起特納氏症候群(Turner's syndrome)。21號染色體的拷貝數增加引起唐氏症候群(Down Syndrome)。侵入性測試(諸如羊膜穿刺術或絨毛膜絨毛取樣(CVS))可導致流產風險,並且此處使用較無創的測試母體血液的方法。
本文所描述的方法可適用於無創檢測胎兒非整倍性。圖12中示出了示例性方法。對處於攜帶具有非整倍體基因組的胎兒的風險中的孕婦進行測試1200。收集含有胎兒基因材料的母體血液樣品1205。然後從血液樣品提取基因材料(例如無細胞核酸)1210。還可獲得條形碼化珠粒的集合1215。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將樣品1210、條形碼化珠粒1220以及(在一些情況下)其他試劑(例如還原劑)組合在一起,並且進行分配。舉例來說,此類分配可涉及將組分引導至微滴產生系統,諸如微流體裝置1225。在微流體裝置1225的輔助下,可形成油包水乳液1230,其中所述乳液含有水性微滴,所述水性微滴含有樣品核酸1210、條形碼化珠粒1215以及(在一些情況下)還原劑。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放1235。隨機N-mer可然後引導樣品核酸的不同區域,從而在擴增之後產生樣品的擴增拷貝,其中將各拷貝用條形碼序列標記1240。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。在其他實施方案中,單個微滴包含獨特的條形碼序列;或者在一些情況下,整個微滴群體中的某一比例具有獨特的序列。隨後,將乳液破壞1245,並且可經由例如擴增方法(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可經由任何適合類型的測序平臺(例如Illumina、Ion Torrent、Pacific Biosciences SMRT、Roche 454測序、SOLiD測序等)進行測序1250,並且應用算法來解釋測序數據1255。測序算法通常能夠例如對條形碼進行分析以比對測序讀段和/或識別特定序列讀段所屬的樣品。可基於所連接的獨特條形碼條形碼將比對序列進一步歸屬於其各自的基因起源(例如,染色體)。然後將染色體拷貝的數目與正常二倍體染色體相比較1260。患者被告知不同染色體的任何拷貝數失常和相關風險/疾病1265。
例如確定基因變體為相連的或位於不同染色體上的定相可為多種應用提供適用信息。舉例來說,定相適用於確定基因組中是否存在與疾病相關的某些易位。此類易位的檢測還可允許鑑別診斷和改良的治療。確定基因組中哪些等位基因是相連的可適用於考慮基因是如何遺傳的。
知道染色體對中的各個單個染色體的等位基因的模式、單倍型經常可能是有用的。舉例來說,存在於一個染色體上的失活突變的兩個拷貝可具有有限的效果,但如果分布在兩個染色體之間就可具有顯著效果,例如在任一染色體提供活性基因產物的情況下。例如在疾病風險增加或對某些藥物的反應缺少的情況下可表現這些效果。
IV.應用方法和系統來識別/表徵結構變異
在其他應用中,本文所描述的方法和系統高度適用於獲得長範圍分子序列信息以識別和表徵廣泛範圍的不同基因結構變異。如上文所提到,這些變異包括多種不同的變體事件,包括插入、缺失、重複、反轉錄轉座子、易位、倒位、短和長串聯重複等。這些結構變異受到大量科學關注,因為它們被認為與一系列多樣的基因疾病相關。
儘管這些變異受到關注,幾乎沒有識別和表徵這些結構變異的有效且高效的方法。這部分是因為這些變異的特徵不是存在異常序列區段,而是實際上涉及將被視為是正常序列區段的異常序列環境,或簡單地丟失序列信息。由於其相對短的讀段長度,大多數測序技術不能提供其所產生的序列讀段的顯著環境,以及尤其例如其讀段長度以外的長範圍序列環境,並且因此損失在組裝過程中對這些變異的識別。識別這些變異時的困難因這些技術的整體方法而進一步複雜化,其中將許多分子(例如多個染色體)組合以產生共有序列,所述共有序列可包括包括與不包括變異的基因組材料。
然而,在當前所描述的方法和系統的情形下,可利用短讀段測序技術來得到長範圍序列信息,所述長範圍序列信息可歸屬於單個起源核酸分子,並且因此保留整個或部分地含於那些單個分子中的變體區域的長範圍序列環境。
如上文所描述,本文所描述的方法和系統能夠提供長範圍序列信息,所述長範圍序列信息可歸屬於單個起源核酸分子,並且此外,在處理此長範圍序列信息時,通過這些更長序列信息的比較和重疊推測甚至更長範圍的序列環境。此類長範圍序列信息和/或推測的序列環境允許識別和表徵使用可獲得的技術不能輕易識別的許多結構變異。
雖然以簡化的方式在圖2中進行了說明,但圖13A和13B提供更詳細的使用本文所描述的方法和系統識別某些類型的結構變異的示例性方法。如圖所示,生物體的基因組或生物體的組織一般會包括圖13A中所說明的第一基因型,其中將包括第一基因1304的第一基因區域1302與包括第二基因1208的第二基因區域1306分離。此分離可反映基因之間的一系列距離,包括例如同一外顯子中的不同區域、同一染色體上的不同外顯子、不同染色體等。然而,如圖13B中所示,示出了一個基因型,所述基因型反映易位事件已發生,其中基因1308被插入基因區域1304中,使得其在基因1304與1308之間形成如變體序列1314中的基因融合1312的基因融合。
當前用於檢測大基因組結構變體(諸如大的倒位或易位)的方法依賴於讀段對,所述讀段對跨越變體的斷點(例如其中易位部分融合在一起的基因組基因座)。為確保在測序實驗期間觀察到此類讀段對,可能需要非常深入的測序。在靶向測序(諸如外顯子組測序)中,使用當前測序技術檢測結構變體幾乎是不可能的,除非斷點在靶向的區域內(例如在外顯子中),這是非常不可能的。
然而,本文所描述的條形碼方法和系統所提供的信息可極大地提高檢測結構變體的能力。直觀上,在斷點左側和右側的基因座,可傾向於位於基因組DNA的共同片段上,並且因此保持在單一分區內,並且因此用共同或共有條形碼序列條形碼化。由於剪切的隨機性質,這種條形碼共有隨著序列距離斷點更遠而減少。使用統計方法,可確定兩個基因組基因座之間的條形碼重疊是否顯著大於偶然將預料的情況。此類重疊暗示斷點的存在。重要地,條形碼信息補充由傳統測序提供的信息(諸如來自跨越斷點的讀段的信息),如果此類信息是可獲得的。
在本文所描述的方法的情況下,如上文所描述,將來自生物體的基因組材料(包括相關基因區域)片段化,使得其包括相對長的片段。這是相對於圖13A中的非易位基因型來說明。如圖所示,形成分別包括基因區域1302和1306的兩個長單個第一分子片段1316和1318。分別將這些片段單獨分配至分區1320和1322中,並且將第一片段中的每一者分別在分區內片段化成許多第二片段1324和1326,此片段化過程將獨特標識標籤或條形碼序列連接至第二片段,所述獨特標識標籤或條形碼序列對給定分區內的所有第二片段來說是共同的。對於分區1320和1322中的每一者,標籤或條形碼分別由「1」或「2」表示。因此,完全分開的基因1304和1308可產生第二片段的區別分配和區別條形碼化群組。
一旦條形碼化,然後即可匯集第二片段並且進行核酸測序過程,所述核酸測序過程可提供第二片段的序列以及所述片段的條形碼序列。基於特定條形碼(例如1或2)的存在,然後如由條形碼歸屬於各序列所示可將第二片段序列歸屬於某一起源序列,例如基因1304或1308。在一些情況下,條形碼化的第二片段序列關於單獨的起源第一片段序列的映射可足夠明確確定未發生易位。然而,在一些情況下,可組裝第二片段序列以提供全部或一部分起源第一片段序列的例如如由組裝序列1330和1332所示的組裝序列。
與圖13A中所示的非易位基因型實例相比之下,圖13B示出了相同方法應用於含有易位的基因型的示意性說明。如圖所示,由變體序列1314產生第一長核酸片段1352,並且包括至少一部分的易位變體,例如基因融合1312。然後將第一片段1352分配至離散分區1354中。在分區1354內,將第一片段1352進一步片段成第二片段1356,所述第二片段又包括獨特條形碼,所述獨特條形碼對於分區1354內的所有第二片段1356來說是相同的(以條形碼「1」示出)。如上所述,匯集第二片段並且測序提供第二片段以及其相關條形碼的基礎序列。然後可將這些條形碼化序列歸屬於其各自的基因序列。然而,如圖所示,兩種基因均可反映包括相同條形碼序列的經歸屬的第二片段序列,表明其起源於相同分區,並且潛在地起源於相同起源分子,從而指示基因融合。這可通過提供許多重疊的第一片段來進一步驗證,所述許多重疊的第一片段也包括基因融合的至少諸多個部分,但在不同分區中用不同條形碼加以處理。
在一些情況下,存在歸屬於最初分離的基因中的每一者的多個不同條形碼序列(和其基礎片段序列)可指示存在基因融合或其他易位事件。在一些情況下,將至少2個條形碼、至少3個不同條形碼、至少4個不同條形碼、至少5個不同條形碼、至少10個不同條形碼、至少20個不同條形碼或更多歸屬於基於參考序列將被預期已分離的兩個基因區域可提供易位事件已將那些區域放置於彼此近端、附近或以其他方式使彼此整合的指示。在一些情況下,被分配的片段的尺寸可指示可識別變體聯繫的靈敏性。特定而言,在給定微滴中的片段的長度為10kb的情況下,將預期在所述10kb尺寸範圍內的所述聯繫將為可檢測的。
同樣地,在變體與野生型結構均在相同的10kb片段內的情況下,將預期所述變體的識別會更困難,因為通過共同或共有條形碼兩者均將顯示聯繫。因此,可使用片段尺寸選擇來調節所檢測的相連序列的相對接近度,無論是野生型還是變體。然而,一般來說,在本文中可通過識別變體基因組中那些不相連的序列區段之間的聯繫而輕易識別產生一般由超過100個鹼基、超過500個鹼基、超過1kb、10kb、超過20kb、超過30kb、超過40kb、超過50kb、超過60kb、超過70kb、超過80kb、超過90kb、超過100kb、超過200kb或甚至更大距離的隔開的近端序列的結構變體,所述聯繫是由共有或共同的條形碼和/或如所提到的由跨越斷點的序列數據指示。當那些相連序列在基因組序列內隔開小於50kb、小於40kb、小於30kb、小於20kb、小於10kb、小於5kb、小於4kb、小於3kb、小於2kb、小於1kb、小於500個鹼基、小於200個鹼基或甚至更小距離時,此類聯繫通常是可識別的。
在一些情況下,產生位於彼此近端或相連的兩個序列(其中它們通常會隔開例如超過10kb、超過20kb、超過30kb、超過40kb或超過50kb或更多)的結構變異可通過佔可映射條形碼化序列的總數的百分比來識別,所述可映射條形碼化序列包括條形碼,所述條形碼對於兩個序列部分來說為共同的。
如應了解,在一些情況下,本文所描述的方法可確保在某一序列距離內的序列(無論是野生型還是變體序列)將被包括在單一分區內,例如作為單一核酸片段。舉例來說,在共同或重疊條形碼序列為映射至兩個序列的條形碼的總數的大於1%的情況下,其可用於識別兩個序列區段之間並且特別是一般將不相連的兩個序列區段之間的聯繫,例如結構變異。在一些情況下,共有或共同條形碼可佔可映射至兩個一般分開的序列的全部條形碼的超過2%、超過3%、超過4%、超過5%、超過6%、超過7%、超過8%並且在一些情況下超過9%或甚至超過10%,以識別構成基因組內的結構變異的結構聯繫。在一些情況下,可以統計顯著大於已知不具有結構變異的對照基因組的比例或數目檢測到共有或共同條形碼。另外,在第二序列片段跨越變體序列遇到「正常」序列的點或「斷點」(例如如在第二片段1358中)的情況下,可使用此信息作為基因融合的額外證據。
再次,如上所述,可通過組裝第二片段序列以產生基因融合1312的組裝序列(以組裝序列1360示出)來進一步闡明基因融合1312的結構。
此外,雖然條形碼序列的存在允許將短序列組裝成更長起源片段的序列,但這些更長片段也允許從由不同的重疊起源長片段組裝的重疊長片段推測更長範圍序列信息。此所得組裝允許基因融合1312的更長範圍序列水平識別和表徵。
在一些情況下,上文所描述的方法適用於識別反轉錄轉座子的存在。可通過剪接信使RNA(mRNA)的轉錄繼之以反轉錄以及插入基因組中的新位置來形成反轉錄轉座子。因此,這些結構變體缺少內含子並且經常為染色體間的,但以其他方式具有多樣的特徵。當反轉錄轉座子引入基因的功能拷貝時,它們被稱為反轉錄基因(retrogene),所述反轉錄基因在人和果蠅(Drosophila)基因組中已有報告。在其他情況下,反轉錄拷貝可含有整個轉錄物、特定轉錄物同種型或不完整轉錄物。另外,替代轉錄起始位點和啟動子序列有時位於轉錄物內,所以反轉錄轉座子有時在基因組的再插入區域內引入啟動子序列,這會驅使下遊序列的表達。
不像串聯重複,反轉錄轉座子遠離親代基因插入外顯子或內含子內。當在基因附近插入時,反轉錄轉座子可利用就近調控序列來進行表達。在基因附近插入還可使接收基因失活或形成新的嵌合體轉錄物。反轉錄轉座子介導的嵌合基因轉錄物在人樣品的RNA-Seq數據中已有報告。
儘管反轉錄轉座子具重要意義,但其檢測可能被限於定向方法,所述定向方法依賴於來自mate pair文庫的成對讀段支持、全基因組測序(WGS)中的外顯子-外顯子接合發現或反轉錄轉座子嵌合體的RNA-Seq識別。所有這些方法均可能具有使分析複雜化的假陽性。
可使用本文所描述的系統和方法從全基因組文庫識別反轉錄轉座子,並且可使用上文所論述的條形碼映射來定位其插入位點。舉例來說,Ceph NA12878基因組具有SKA3-DDX10嵌合反轉錄轉座子。SKA3無內含子轉錄物被插入DDX10的外顯子10與11之間。此外,還可使用本文所描述的方法檢測NA12878中的CBX3-C15ORF17反轉錄轉座子。CBX3的同種型2被插入C15ORF17的外顯子2與3之間。已在20%的來自HapMap計劃的歐洲RNA-Seq樣品中觀測到此嵌合轉錄物(D.R.Schrider等PLoS Genetics 2013)。
還可使用本文所描述的方法和系統在所製備的全外顯子組文庫中檢測反轉錄轉座子。雖然在外顯子組靶向的情況下反轉錄轉座子容易富集,但可能難以或不可能區分易位事件和反轉錄轉座子,因為內含子在捕獲過程中被去除。然而,使用本文所描述的系統和方法,通過為疑似反轉錄轉座子引入內含子誘餌可在全外顯子組測序(WES)文庫中識別反轉錄轉座子(也參見2014年10月29日提交的美國臨時專利申請號62/072,164,該臨時專利申請出於所有目的以全文引用的方式併入本文中)。缺少內含子信號可指示反轉錄轉座子結構變體,而內含子信號可指示易位。
如應了解,在識別和表徵上文所描述的變異時使用更長範圍序列環境的能力同樣適用於通過將條形碼映射至變異內和/或跨越變異的區域來識別其他結構變異的範圍,包括插入、缺失、反轉錄轉座子、倒位等。
V.由拷貝數變異引起的疾病和病症
本發明方法和系統提供高度準確和靈敏的用於診斷和/或檢測廣泛範圍的疾病和病症的方法。與拷貝數變異相關的疾病可包括例如迪喬治/顎心面症候群(DiGeorge/velocardiofacial syndrome)(22q11.2缺失)、普拉德-威利症候群(Prader-Willi syndrome)(15q11-q13缺失)、威廉-博伊倫症候群(Williams-Beuren syndrome)(7q11.23缺失)、米勒-狄克症候群(Miller-Dieker syndrome)(MDLS)(17p13.3微缺失)、史密斯-馬吉利斯症候群(Smith-Magenis syndrome)(SMS)(17p11.2微缺失)、神經纖維瘤病1型(NF1)(17q11.2微缺失)、費倫-麥克德米德症候群(Phelan-McErmid Syndrome)(22q13缺失)、雷特症候群(Rett syndrome)(染色體Xq28上的MECp2的功能缺失突變)、梅茨巴赫病(Merzbacher disease)(PLP1的CNV)、脊髓性肌萎縮(SMA)(染色體5q13上端粒SMN1的純合性不存在)、波託茨基-魯普斯基症候群(Potocki-Lupski Syndrome)(PTLS,染色體17p.11.2重複)。PMP22基因的額外拷貝可與沙-馬-圖神經病變IA型(Charcot-Marie-Tooth neuropathy type IA,CMT1A)和遺傳性壓力易感性神經病變(hereditary neuropathy with liability to pressure palsies,HNPP)相關。所述疾病可為描述於Lupski J.(2007)Nature Genetics 39:S43-S47中的疾病。
本文所提供的方法和系統還可以準確檢測或診斷廣泛範圍的胎兒非整倍性。經常,本文所提供的方法包括分析從孕婦取得的樣品(例如血液樣品)以評估樣品內的胎兒核酸。胎兒非整倍性可包括例如13三體(帕韜氏症候群(Patau syndrome))、18三體(愛德華茲症候群(Edwards syndrome))、21三體(唐氏症候群)、柯林菲特氏症(Klinefelter Syndrome)(XXY)、一個或多個染色體的單體性(X染色體單體性,特納氏症候群)、X三體性、一個或多個染色體的三體性、一個或多個染色體的四體性或五體性(例如XXXX、XXYY、XXXY、XYYY、XXXXX、XXXXY、XXXYY、XYYYY以及XXYYY)、三倍性(每個染色體有三個,例如人中的69個染色體)、四倍性(每個染色體有四個,例如人中的92個染色體)以及多倍性。在一些實施方案中,非整倍性可為區段非整倍性。區段非整倍性可包括例如1p36重複、dup(17)(p11.2p11.2)症候群、唐氏症候群、佩利措伊斯-梅茨巴赫病(Pelizaeus-Merzbacher disease)、dup(22)(q11.2q11.2)症候群以及貓眼症候群。在一些情況下,異常基因型(例如胎兒基因型)是歸因於性染色體或常染色體的一個或多個缺失,此可導致諸如以下病症:貓叫症候群(Cri-du-chat syndrome)、沃夫-賀許宏氏症(Wolf-Hirschhorn)、威廉-博伊倫症候群、沙-馬-圖病(Charcot-Marie-Tooth disease)、遺傳性壓力易感性神經病變、史密斯-馬吉利斯症候群、神經纖維瘤病、阿拉吉耶症候群(Alagille syndrome)、顎心面症候群(Velocardiofacial syndrome)、迪喬治症候群(DiGeorgesyndrome)、類固醇硫酸酯酶缺乏症、卡曼氏症候群(Kallmann syndrome)、小眼球線性皮膚缺損、腎上腺發育不良、甘油激酶缺乏症、佩利措伊斯-梅茨巴赫病、Y上睪丸決定因子、無精症(因子a)、無精症(因子b)、無精症(因子c)或1p36缺失。在一些實施方案中,染色體數目減少導致XO症候群。
過度基因組DNA拷貝數變異也與李-佛美尼癌症傾向症候群(Li-Fraumeni cancer predisposition syndrome)相關(Shlien等(2008)PNAS105:11264-9)。CNV與畸形症候群相關,包括CHARGE(眼部缺損、心臟異常、後鼻孔閉鎖、發育遲緩、生殖器以及耳部異常)、彼得斯-普拉斯症候群(Peters-Plus)、皮特-霍普金斯症候群(Pitt-Hopkins)以及血小板減少-橈骨缺失症候群(thrombocytopenia-absent radius syndrome)(參見例如Ropers HH(2007)Am J of Hum Genetics 81:199-207)。拷貝數變異與癌症之間的關係描述於例如Shlien A.和Malkin D.(2009)Genome Med.1(6):62中。拷貝數變異與例如自閉症、精神分裂症以及特發性學習障礙相關。參見例如Sebat J.等(2007)Science 316:445-9;Pinto J.等。
如本文所描述,本文所提供的方法和系統還適用於檢測與不同類型的癌症相關的CNV。舉例來說,可使用所述方法和系統來檢測EGFR拷貝數,在非小細胞肺癌中其可為增加的。
還可使用本文所提供的方法和系統來測定受試者對特定疾病或病症的易感性水平,包括病原體感染易感性(例如病毒、細菌、微生物、真菌等)。舉例來說,鑑於相對高水平的CCL3L1與更低的HIV感染易感性相關,可使用所述方法通過分析CCL3L1的拷貝數來測定受試者對HIV感染的易感性(Gonzalez E.等(2005)Science 307:1434-1440)。在另一實例中,可使用所述方法來測定受試者對系統性紅斑狼瘡的易感性。在此類情況下,舉例來說,可使用所述方法來檢測FCGR3B(CD16細胞表面免疫球蛋白受體)的拷貝數,因為此分子的低拷貝數與增加的系統性紅斑狼瘡易感性相關(Aitman T.J.等(2006)Nature 439:851-855)。還可使用本文所提供的方法和系統來檢測與其他疾病或病症相關的CNV,諸如與自閉症、精神分裂症以及特發性學習障礙相關的CNV(Kinght等,(1999)TheLancet 354(9191):1676–81)。類似地,可使用所述方法和系統來檢測常染色體顯性小耳症,其與染色體4p16處的拷貝數可變區的五個串聯拷貝有關(Balikova I.(2008)Am J.Hum Genet.82:181-187)。
VI.疾病和病症的檢測、診斷以及治療
本文所提供的方法和系統還可協助檢測、診斷以及治療疾病或病症。在一些情況下,一種方法包括使用本文所描述的系統或方法檢測疾病或病症,並且基於對疾病的檢測進一步為受試者提供治療。舉例來說,如果檢測到癌症,那麼可通過外科手術、通過施用被設計成治療此類癌症的藥物、通過提供激素治療以及/或者通過施用輻射或更一般化的化學治療來治療受試者。
經常,所述方法和系統還允許鑑別診斷並且可進一步包括用靶向治療來治療患者。一般來說,可通過以下方式實現疾病或病症的鑑別診斷(或其不存在):測定和表徵從懷疑患有所述疾病或病症的受試者獲得的樣品核酸的序列,並且通過將其與指示病症或疾病狀態存在(或不存在)的參考核酸的序列和/或序列表徵相比較將樣品核酸進一步表徵為指示病症或疾病狀態(或其不存在)。
參考核酸序列可源於指示疾病或病症狀態不存在的基因組(例如生殖系核酸)或可源於指示疾病或病症狀態的基因組(例如癌症核酸、指示非整倍性等的核酸)。另外,可在一個或多個方面表徵參考核酸序列(例如具有長於1kb、長於5kb、長於10kb、長於15kb、長於20kb、長於30kb、長於40kb、長於50kb、長於60kb、長於70kb、長於80kb、長於90kb或甚至長於100kb的長度),其中非限制性實例包括確定特定序列的存在(或不存在)、確定特定單倍型的存在(或不存在)、確定一種或多種基因變異(例如結構變異(例如拷貝數變異、插入、缺失、易位、倒位、反轉錄轉座子、重排、重複擴增、重複等)、單核苷酸多態性(SNP)等)的存在(或不存在)以及其組合。另外,可使用任何合適類型和數目的參考序列的序列特徵來表徵樣品核酸的序列。舉例來說,可使用參考核酸序列的一種或多種基因變異(或其缺乏)或結構變異(或其缺乏)作為用於將參考核酸識別為指示病症或疾病狀態存在(或不存在)的序列標籤。基於對所利用的參考核酸序列的表徵,可以類似方式表徵樣品核酸序列並且基於其是否展示與參考核酸序列類似的性質將其進一步表徵/識別為源於(或不源於)指示病症或疾病的核酸。在一些情況下,可在經過編程的計算機處理器的輔助下完成樣品核酸序列和/或參考核酸序列的表徵以及其比較。在一些情況下,此類經過編程的計算機處理器可被包括於計算機控制系統中,諸如本文中別處所描述的示例性計算機控制系統中。
可從任何合適的來源獲得樣品核酸,包括本文中別處所描述的樣品來源和生物樣品來源。在一些情況下,樣品核酸可包括無細胞核酸。在一些情況下,樣品核酸可包括腫瘤核酸(例如,腫瘤DNA)。在一些情況下,樣品核酸可包括循環腫瘤核酸(例如,循環腫瘤DNA(ctDNA))。循環腫瘤核酸可源於循環腫瘤細胞(CTC)和/或可從例如受試者的血液、血漿、其他體液或組織獲得。
圖20-21說明用於在疾病檢測和診斷的背景下表徵樣品核酸的示例性方法。圖20展現用於諸如以類似於圖6中所示的方式由更短條形碼化片段測定參考核酸(例如生殖系核酸(例如生殖系基因組DNA)、與特定病症或疾病狀態相關的核酸)的長範圍序列環境的示例性方法。就圖20來說,可獲得參考核酸2000,並且可獲得條形碼化珠粒的集合2010。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將參考核酸2005、條形碼化珠粒、2015以及(在一些情況下)其他試劑(例如還原劑)2020組合併且進行分配。在一些情況下,可在分配之前將參考核酸2000片段化,並且將所得片段中的至少一些如2005進行分配以便進行條形碼化。舉例來說,此類分配可涉及將組分引入微滴產生系統,諸如微流體裝置2025。在微流體裝置2025的輔助下,可形成油包水乳液2030,其中所述乳液含有水性微滴,所述水性微滴含有參考核酸2005、還原劑2020以及條形碼化珠粒2015。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放2035。隨機N-mer可然後引導參考核酸的不同區域,從而在擴增之後產生參考核酸的擴增拷貝,其中將各拷貝用條形碼序列標記2040。在一些情況下,可通過類似於本文中別處所描述並且示意性描繪於圖5中的方法來實現擴增2040。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。隨後,將乳液破壞2045,並且可經由例如擴增方法2050(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可進行測序2055,並且應用算法來解釋測序數據2060。在一些情況下,測序數據的解釋2060可包括提供參考核酸的至少一部分的序列。在一些情況下,獲得參考核酸的長範圍序列環境並且進行表徵,諸如在參考核酸源於疾病狀態的情況下(例如如本文中別處所描述的一個或多個單倍型的測定、一種或多種結構變異(例如拷貝數變異、插入、缺失、易位、倒位、重排、重複擴增、重複、反轉錄轉座子、基因融合等)的測定、一個或多個SNP等的分辨等)。在一些情況下,可針對從來源獲得的各種參考核酸和所產生的推測重疊群來識別變體以提供更長範圍序列環境,諸如本文中別處關於圖7所描述。
圖21展現由如圖20中所示所獲得的參考2060表徵來表徵樣品核酸序列的實例。可如本文中別處所描述諸如經由示意性描繪於圖6中的方法由更短條形碼化片段的測序獲得樣品核酸的長範圍序列環境。如圖21中所示,可從懷疑患有病症或疾病(例如癌症)的受試者獲得核酸樣品(例如包含循環腫瘤核酸的樣品)2100,並且還可獲得條形碼化珠粒2110。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將樣品核酸2105、條形碼化珠粒2115以及(在一些情況下)其他試劑(例如還原劑)2120組合併且進行分配。在一些情況下,在分配之前將胎兒樣品2100片段化並且對所得片段中的至少一些如2105進行分配以便條形碼化。舉例來說,此類分配可涉及將組分引入微滴產生系統(諸如微流體裝置)2125。在微流體裝置2125的輔助下,可形成油包水乳液2130,其中所述乳液含有水性微滴,所述水性微滴含有樣品核酸2105、還原劑2120以及條形碼化珠粒2115。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放2135。隨機N-mer可然後引導樣品核酸的不同區域,從而在擴增之後產生樣品核酸的擴增拷貝,其中將各拷貝用條形碼序列標記2140。在一些情況下,可通過類似於本文中別處所描述並且示意性描繪於圖5中的方法來實現擴增2140。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。隨後,將乳液破壞2145,並且可經由例如擴增方法2150(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可進行測序2155並且應用算法來解釋測序數據2160。在一些情況下,測序數據的解釋2160可包括提供樣品核酸的序列。在一些情況下,獲得核酸樣品的長範圍序列環境。可使用對參考核酸序列的表徵2060來表徵樣品核酸序列2160(例如測定如本文中別處所描述的一個或多個單倍型、測定一種或多種結構變異(例如拷貝數變異、插入、缺失、易位、倒位、重排、重複擴增、重複、反轉錄轉座子、基因融合等)。基於樣品核酸序列及其表徵與參考核酸的序列和表徵的比較,可進行關於病症或疾病狀態的存在(或不存在)的鑑別診斷2170。
如可理解,參考核酸和樣品核酸的分析可作為單獨分配分析來完成或可作為單一分配分析的一部分來完成。舉例來說,可將樣品和參考核酸添加至同一裝置,並且根據圖20和21在微滴中產生條形碼化的樣品和參考片段,其中乳液包含用於兩種類型的核酸的微滴。然後可將乳液破壞,並且匯集微滴的內容物,進一步處理(例如經由PCR批量添加額外序列)並且如本文中別處所描述進行測序。可經由條形碼序列將來自條形碼化片段的單個測序讀段歸屬至其各自的樣品序列。可基於對參考核酸序列的表徵來表徵從樣品核酸獲得的序列。
利用本文的方法和系統可提高測定核酸的長範圍序列環境(包括如本文所描述的參考和樣品核酸序列的長範圍序列環境)的準確度。本文所提供的方法和系統可以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%、或99.999%的準確度測定參考和/或樣品核酸的長範圍序列環境。在一些情況下,本文所提供的方法和系統可以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%、或0.000005%的誤差率測定參考和/或樣品核酸的長範圍序列環境。
另外,本文的方法和系統還可在一個或多個方面(例如序列的測定、一種或多種基因變異的測定、單倍型的測定等)提高表徵參考核酸序列和/或樣品核酸序列時的準確度。因此,本文所提供的方法和系統可以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%、或99.999%的準確度在一個或多個方面表徵參考核酸序列和/或樣品核酸序列。在一些情況下,本文所提供的方法和系統可以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%、或0.000005%的誤差率在一個或多個方面表徵參考核酸序列和/或樣品核酸序列。
另外,如上文所論述,測定參考核酸的長範圍序列環境和對其進行表徵時的準確度提高可使得對樣品核酸進行測序和表徵以及隨後用於鑑別診斷病症或疾病時的準確度提高。因此,可以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%、或0.000005%的誤差率由參考核酸序列的分析提供樣品核酸序列(包括長範圍序列環境)。在一些情況下,可通過與參考核酸的序列和/或序列表徵相比較使用樣品核酸序列以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%、或99.999%的準確度來鑑別診斷病症或疾病(或其不存在)。在一些情況下,可通過與參考核酸的序列和/或序列表徵相比較使用樣品核酸序列以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%、或0.000005%的誤差率來鑑別診斷病症或疾病(或其不存在)。
在一個實例中,可使用所述方法和系統來檢測肺癌患者的拷貝數變異以確定肺癌是否是與EGFR基因變異相關的非小細胞肺癌。在此類診斷之後,可細化患者的治療方案以與鑑別診斷相關聯。靶向治療或分子靶向治療是癌症醫學治療(藥物治療)的主要形式之一,其他是激素治療和細胞毒性化學治療。靶向治療通過幹擾為致癌作用和腫瘤生長所需的特定靶向分子,而不是通過簡單地幹擾所有快速分裂的細胞(例如在傳統化學治療情況下)來阻礙癌細胞生長。
圖14示出了鑑別診斷非小細胞肺癌的示例性方法。對慢性咳嗽、體重減輕以及呼吸急促患者進行關於肺癌的測試1400。從患者抽取血液1405,並且從血液得到樣品(例如循環腫瘤細胞、無細胞DNA、循環核酸(例如循環腫瘤核酸)等)1410。還可獲得條形碼化珠粒的集合1415。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將樣品1410、條形碼化珠粒1420以及(在一些情況下)其他試劑(例如還原劑)組合併且進行分配。舉例來說,此類分配可涉及將組分引入微滴產生系統,諸如微流體裝置1425。在微流體裝置1425的輔助下,可形成油包水乳液1430,其中所述乳液含有水性微滴,所述水性微滴含有樣品核酸1410、條形碼化珠粒1415以及(在一些情況下)還原劑。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放1435。隨機N-mer可然後引導樣品核酸的不同區域,從而在擴增之後產生樣品的擴增拷貝,其中將各拷貝用條形碼序列標記1440。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。隨後,將乳液破壞1445,並且可經由例如擴增方法(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可進行測序1450,並且應用算法來解釋測序數據1455。測序算法通常能夠例如對條形碼進行分析以比對測序讀段和/或識別特定序列讀段所屬的樣品。
然後將分析過的序列與已知基因組參考序列相比較以確定不同基因的CNV 1460。如果DNA中的EGFR拷貝數高於正常,那麼可將患者鑑別診斷為患有非小細胞肺癌(NSCLC)而不是小細胞肺癌1465。非小細胞肺癌的CTC還具有其他拷貝數變異,所述其他拷貝數變異可使其與小細胞肺癌進一步區分開。視癌症的階段而定,囑咐進行手術、化學治療或放射治療1470。在一些情況下,為被診斷為患有NSLC的患者施用針對此類癌症的藥物,諸如ALK抑制劑(例如克唑替尼(Crizotinib))。在EGFR的變異的一些情況下,為患者施用西妥昔單抗(cetuximab)、帕尼單抗(panitumumab)、拉帕替尼(lapatinib)以及/或者卡培他濱(capecitabine)。在不同的情況下,目標可為不同基因,諸如ERBB2,並且治療包括曲妥珠單抗(trastuzumab)(赫賽汀(Herceptin))。(2010)Nature466:368-72;CookE.H.和Scherer S.W.(2008)Nature 455:919-923。
靶向治療的主要類別為小分子、小分子藥物綴合物以及單克隆抗體。小分子可包括酪氨酸激酶抑制劑,諸如伊馬替尼(Imatinib)甲磺酸鹽(格列衛(Gleevec),也被稱為STI–571)(其被批准用於慢性粒細胞性白血病、胃腸道基質腫瘤以及一些其他類型的癌症);吉非替尼(Gefitinib)(易瑞沙(Iressa),也被稱為ZD1839)(其靶向表皮生長因子受體(EGFR)酪氨酸激酶並且在美國被批准用於非小細胞肺癌);厄洛替尼(Erlotinib)(以特羅凱(Tarceva)形式出售);硼替佐米(Bortezomib)(萬珂(Velcade))(其為誘導細胞凋亡的蛋白酶體抑制劑藥物,其通過幹擾蛋白質而使得癌細胞經歷細胞死亡);他莫昔芬(tamoxifen);JAK抑制劑(例如託法替尼(tofactinib))、ALK抑制劑(例如克唑替尼);Bcl-2抑制劑(例如在臨床試驗中的奧巴克拉(obatoclax)、ABT-263以及棉酚(Gossypol));PARP抑制劑(例如依尼帕尼(Iniparib)、在臨床試驗中的奧拉帕尼(Olaparib));PI3K抑制劑(例如在III期試驗中的哌立福辛(perifosine))。阿帕替尼(其為選擇性VEGF受體2抑制劑);AN-152,(AEZS-108)與[D-Lys(6)]-LHRH鍵聯的多柔比星(doxorubicin);Braf抑制劑(維羅非尼(vemurafenib)、達拉菲尼(dabrafenib)、LGX818)(用於治療具有BRAF V600E突變的轉移性黑色素瘤);MEK抑制劑(曲美替尼(trametinib),MEK162);CDK抑制劑,例如PD-0332991、在臨床試驗中的LEE011;Hsp90抑制劑;以及沙利黴素(Salinomycin)。
其他治療包括小分子藥物綴合物,諸如Vintafolide,其為由靶向葉酸受體的小分子組成的小分子藥物綴合物。
單克隆抗體為另一類型的治療,其可作為本文所提供的方法的一部分進行施用。還可施用單克隆藥物綴合物。示例性單克隆抗體包括:利妥昔單抗(Rituximab)(以MabThera或Rituxan出售)(其靶向存在於B細胞上的CD20,並且靶向非霍奇金淋巴瘤(non Hodgkin lymphoma));曲妥珠單抗(赫賽汀)(其靶向在一些類型的乳房癌中表達的Her2/neu(也稱為ErbB2)受體);西妥昔單抗(以(Erbitux出售)以及帕尼單抗貝伐單抗(Bevacizumab)(以Avastin形式出售)(其靶向VEGF配位體)。
VII.由親代核酸表徵胎兒核酸
如本文中別處所提到,還可使用本文所描述的方法和系統來表徵受試者的血液或血漿內的循環核酸。此類分析包括分析循環腫瘤DNA,以便用於識別患者中的潛在疾病狀態;或懷孕女性的血液或血漿內的循環胎兒DNA,以例如在沒有通過羊膜穿刺術或其他侵入性程序進行直接取樣的情況下按無創的方式表徵胎兒DNA。
在一些情況下,可至少部分基於親代核酸序列的分析使用所述方法來表徵胎兒核酸序列,例如循環胎兒DNA。舉例來說,可使用本文所描述的方法和系統由更短條形碼化片段測定父體與母體核酸的長範圍序列環境(例如具有長於1kb、長於5kb、長於10kb、長於15kb、長於20kb、長於30kb、長於40kb、長於50kb、長於60kb、長於70kb、長於80kb、長於90kb或甚至長於100kb的長度)。可使用長範圍序列環境來測定一個或多個單倍型和一種或多種基因變異,包括父體與母體核酸序列中的單核苷酸多態性(SNP)、結構變異(例如拷貝數變異、插入、缺失、易位、倒位、重排、重複擴增、反轉錄轉座子、重複、基因融合等)。另外,可使用父體和母體核酸的長範圍序列環境以及任何所測定的SNP、單倍型和/或結構變異信息來表徵從懷孕母親獲得的胎兒核酸(例如循環胎兒核酸,諸如無細胞胎兒核酸)的序列。在一些情況下,經由與母體和父體序列和表徵相比較來表徵胎兒核酸可在經過編程的計算機處理器的輔助下完成。在一些情況下,此類經過編程的計算機處理器可被包括於計算機控制系統中,諸如本文中別處所描述的示例性計算機控制系統中。
舉例來說,可使用親代和/或母體核酸的序列和/或長範圍序列環境作為用於表徵胎兒核酸(包括胎兒核酸序列)的參考。事實上,由本文所描述的方法和系統獲得的長範圍序列環境可提供父體和母體核酸的改善的長範圍序列環境信息,由此可表徵胎兒核酸序列。在一些情況下,由親代核酸作為參考來表徵胎兒核酸序列可包括測定胎兒核酸的至少一部分的序列,以及/或者識別胎兒核酸序列的一個或多個SNP,測定胎兒核酸序列的一個或多個從頭突變,測定胎兒核酸序列的一個或多個單倍型,以及/或者測定和表徵胎兒核酸序列中的一種或多種結構變異等。
圖17-19說明經由對更短條形碼化片段進行測序由針對父體和母體核酸所獲得的更長範圍序列環境表徵胎兒核酸的示例性方法。圖17展現可用於諸如以類似於圖6中所示的方法由更短條形碼化片段測定父體核酸樣品(例如父體基因組DNA)的更長範圍序列環境的示例性方法。就圖17來說,可從胎兒的父親獲得包含父體核酸的樣品1700,並且還可獲得條形碼化珠粒集合1710。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將包含核酸的父體樣品1705、條形碼化珠粒1715以及(在一些情況下)其他試劑(例如還原劑)1720組合併且進行分配。在一些情況下,在分配之前將父體樣品1700片段化,並且對所得片段中的至少一些如1705進行分配以便進行條形碼化。舉例來說,此類分配可涉及將組分引入微滴產生系統,諸如微流體裝置1725。在微流體裝置1725的輔助下,可形成油包水乳液1730,其中所述乳液含有水性微滴,所述水性微滴含有父體樣品核酸1705、還原劑1720以及條形碼化珠粒1715。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放1735。隨機N-mer可然後引導父體樣品核酸的不同區域,從而在擴增之後產生父體樣品的擴增拷貝,其中將各拷貝用條形碼序列標記1740。在一些情況下,可通過類似於本文中別處所描述並且示意性描繪於圖5中的方法來實現擴增1740。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。隨後,將乳液破壞1745,並且可經由例如擴增方法1750(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可進行測序1755,並且應用算法來解釋測序數據1760。在一些情況下,舉例來說,測序數據的解釋1760可包括提供父體核酸的至少一部分的序列。在一些情況下,可獲得父體核酸樣品的長範圍序列環境並且進行表徵(例如測定如本文中別處所描述的一個或多個單倍型、測定一種或多種結構變異(例如拷貝數變異、插入、缺失、易位、倒位、重排、重複擴增、重複、反轉錄轉座子、基因融合等)、識別一個或多個SNP、測定一種或多種其他基因變異等)。在一些情況下,可針對各種父體核酸和所產生的推測重疊群來識別變體以提供更長範圍序列環境,諸如本文中別處關於圖7所描述。
圖18展現可用於諸如以類似於圖6中所示的方法由更短條形碼化片段測定母體核酸樣品(例如母體基因組DNA)的長範圍序列環境的示例性方法。就圖18來說,可從胎兒的懷孕母親獲得包含母體核酸的樣品1800,並且還可獲得條形碼化珠粒1810。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將包含核酸的母體樣品1805、條形碼化珠粒1815以及(在一些情況下)其他試劑(例如還原劑)1820組合併且進行分配。在一些情況下,在分配之前將母體樣品1800片段化並且對所得片段中的至少一些如1805進行分配以便進行條形碼化。舉例來說,此類分配可涉及將組分引入微滴產生系統,諸如微流體裝置1825。在微流體裝置1825的輔助下,可形成油包水乳液1830,其中所述乳液含有水性微滴,所述水性微滴含有母體樣品核酸1805、還原劑1820以及條形碼化珠粒1815。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放1835。隨機N-mer可然後引導母體樣品核酸的不同區域,從而在擴增之後產生母體樣品的擴增拷貝,其中將各拷貝用條形碼序列標記1840。在一些情況下,可通過類似於本文中別處所描述並且示意性展示於圖5中的方法來實現擴增1840。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。隨後,將乳液破壞1845,並且可經由例如擴增方法1850(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可進行測序1855,並且應用算法來解釋測序數據1860。在一些情況下,舉例來說,測序數據的解釋1860可包括提供母體核酸的至少一部分的序列。在一些情況下,可獲得母體核酸樣品的長範圍序列環境並且進行表徵(例如測定如本文中別處所描述的一個或多個單倍型、測定一種或多種結構變異(例如拷貝數變異、插入、缺失、易位、倒位、重排、重複擴增、重複、反轉錄轉座子、基因融合等)、識別一個或多個SNP、測定一個或多個其他基因變異等。在一些情況下,可針對從樣品獲得的各種母體核酸和所產生的推測重疊群來識別變體以提供更長範圍序列環境,諸如本文中別處關於圖7所描述。
圖19展現由分別如圖17和圖18中所示所獲得父體1760和母體1860表徵來表徵胎兒樣品序列的實例。如圖19中所示,可從懷孕母親獲得胎兒核酸樣品1900。可如本文中別處所描述諸如經由示意性描繪於圖6中的方法由更短條形碼化片段的測序獲得胎兒核酸的長範圍序列環境。在一些情況下,胎兒核酸樣品可為循環胎兒DNA和/或無細胞DNA,其可例如從懷孕母親的血液、血漿、其他身體流體或組織獲得。還可獲得條形碼化珠粒的集合1910。可使珠粒鍵聯至含有一個或多個條形碼序列以及引物(諸如隨機N-mer或其他引物)的寡核苷酸。在一些情況下,條形碼序列可從條形碼化珠粒釋放,例如通過在條形碼與珠粒之間的鍵的裂解或通過下面珠粒的降解來釋放條形碼,或兩種途徑的組合。舉例來說,在一些方面,條形碼化珠粒可由諸如還原劑等試劑降解或溶解以釋放條形碼序列。在此實例中,將包含核酸的胎兒樣品1905、條形碼化珠粒1915以及(在一些情況下)其他試劑(例如還原劑)1920組合併且進行分配1905.在一些情況下,在分配之前將胎兒樣品1900片段化並且對所得片段中的至少一些如1905進行分配以便進行條形碼化。舉例來說,此類分配可涉及將組分引入微滴產生系統,諸如微流體裝置1925。在微流體裝置1925的輔助下,可形成油包水乳液1930,其中所述乳液含有水性微滴,所述水性微滴含有母體樣品核酸1905、還原劑1920以及條形碼化珠粒1915。還原劑可溶解或降解條形碼化珠粒,由此使具有條形碼和隨機N-mer的寡核苷酸從微滴內的珠粒釋放1935。隨機N-mer可然後引導胎兒樣品核酸的不同區域,從而在擴增之後產生胎兒樣品的擴增拷貝,其中將各拷貝用條形碼序列標記1940。在一些情況下,可通過類似於本文中別處所描述並且示意性描繪於圖5中的方法來實現擴增1940。在一些情況下,各微滴含有寡核苷酸的集合,所述寡核苷酸的集合含有相同的條形碼序列和不同的隨機N-mer序列。隨後,將乳液破壞1945,並且可經由例如擴增方法1950(例如PCR)來添加額外序列(例如輔助特定測序方法的序列、額外條形碼等)。然後可進行測序1955,並且應用算法來解釋測序數據1960。一般來說,可從測序過的更短條形碼化片段獲得胎兒核酸樣品的更長範圍序列環境。在一些情況下,舉例來說,測序數據的解釋1960可包括提供胎兒核酸的至少一部分的序列。可使用父體1760和母體1860樣品的長範圍序列環境和/或表徵來表徵胎兒核酸序列1960(例如測定如本文中別處所描述的一個或多個單倍型、測定一種或多種結構變異(例如拷貝數變異、插入、缺失、易位、倒位、重排、重複擴增、重複、反轉錄轉座子、基因融合等)、測定一個或多個從頭突變、識別一個或多個SNP等)。在一些情況下,可通過將胎兒核酸序列與母體和父體相位區塊相比較來測定胎兒核酸的相位區塊。
如可理解,父體核酸、母體核酸以及/或者胎兒核酸的分析可作為單獨分配分析的一部分來完成或可作為一個或多個組合分配分析的一部分來完成。舉例來說,可將父體、母體以及胎兒核酸添加至同一裝置,並且根據圖17-19在微滴中產生條形碼化母體、父體以及胎兒片段,其中乳液包含用於三種類型的核酸的微滴。然後可將乳液破壞,並且匯集微滴的內容物,進一步處理(例如經由PCR批量添加額外序列)並且如本文中別處所描述進行測序。可經由條形碼序列將來自條形碼化片段的單個測序讀段歸屬至其各自的樣品序列。
在一些情況下,可由使用本文所描述的方法和系統獲得的長範圍父體和母體序列環境和表徵測定胎兒核酸的序列(包括胎兒基因組的序列)和/或胎兒核酸序列中的基因變異。舉例來說,可使用父體和母體基因組的基因組測序以及循環胎兒核酸的測序來測定相應的胎兒基因組序列。由親代基因組和無細胞胎兒核酸的序列分析測定基因組胎兒核酸的序列的實例可見於Kitzman等(2012年6月6日)Sci Transl.Med.4(137):137ra76中,該文獻以引用的方式全部併入本文中。胎兒基因組的測定可適用於胎兒的基因病症(包括例如胎兒非整倍性)的產前確定和診斷。如本文中別處所論述,本文所提供的方法和系統可適用於解析核酸序列中的單倍型。可分別測定父體和母體樣品核酸序列的單倍型解析型父體和母體序列,其可輔助更準確測定胎兒基因組的序列和/或對其進行表徵。
利用本文的方法和系統可提高測定核酸的長範圍序列環境(包括親代核酸序列(例如母體核酸序列、父體核酸序列)的長範圍序列環境)時的準確度。本文所提供的方法和系統可以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%或99.999%的準確度測定親代核酸的長範圍序列環境。在一些情況下,本文所提供的方法和系統可以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%或0.000005%的誤差率測定親代核酸的長範圍序列環境。另外,本文的方法和系統還可在一個或多個方面(例如序列的測定、一種或多種基因變異的測定、一個或多個結構變體的測定、單倍型的測定等)提高表徵父體核酸序列時的準確度。因此,本文所提供的方法和系統可以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%或99.999%準確度在一個或多個方面表徵父體核酸序列。在一些情況下,本文所提供的方法和系統可以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%或0.000005%的誤差率在一個或多個方面表徵親代核酸序列。
另外,如上文所論述,測定親代核酸的長範圍序列環境和對其進行表徵時的準確度提高可使得對胎兒核酸進行測序和表徵時的準確度提高。因此,在一些情況下,可由親代核酸序列的分析以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%或99.999%的準確度提供胎兒核酸序列(包括長範圍序列環境)。在一些情況下,可由親代核酸序列的分析以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%或0.000005%的誤差率提供胎兒核酸序列(包括長範圍序列環境)。在一些情況下,可以至少70%、80%、85%、90%、91%、92%、93%、94%、95%、99%、99.1%、99.2%、99.3%99.4%、99.5%、99.6%、99.7%、99.8%、99.9%、99.95%、99.99%、99.995%或99.999%的準確度在一個或多個方面經由如本文所描述的親代核酸序列的分析(例如序列的測定、一種或多種基因變異的測定、一種或多種結構變異的測定、單倍型的測定等)表徵胎兒核酸序列。在一些情況下,可以低於10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.1%、0.05%、0.01%、0.005%、0.001%、0.0005%、0.0001%、0.00005%、0.00001%或0.000005%的誤差率在一個或多個方面經由如本文所描述的親代核酸序列的分析(例如序列的測定、一種或多種基因變異的測定、單倍型的測定、一種或多種結構變異的測定等)表徵胎兒核酸序列。
VIII.樣品
可以從患者獲得樣品來開始疾病或病症的檢測。如本文中所用,術語「樣品」通常是指生物樣品。生物樣品的實例包括核酸分子、胺基酸、多肽、蛋白質、碳水化合物、脂肪或病毒。在一個實例中,生物樣品為包括一個或多個核酸分子的核酸樣品。示例性樣品可包括聚核苷酸、核酸、寡核苷酸、無細胞核酸(例如無細胞DNA(cfDNA))、循環無細胞核酸、循環腫瘤核酸(例如循環腫瘤DNA(ctDNA))、循環腫瘤細胞(CTC)核酸、核酸片段、核苷酸、DNA、RNA、肽聚核苷酸、互補DNA(cDNA)、雙鏈DNA(dsDNA)、單鏈DNA(ssDNA)、質粒DNA、粘粒DNA、染色體DNA、基因組DNA(gDNA)、病毒DNA、細菌DNA、mtDNA(線粒體DNA)、核糖體RNA、無細胞DNA、無細胞胎兒DNA(cffDNA)、mRNA、rRNA、tRNA、nRNA、siRNA、snRNA、snoRNA、scaRNA、微RNA、dsRNA、病毒RNA等。總之,所用的樣品可視特定處理需要而變化。
包含核酸的任何物質均可為樣品的來源。物質可為流體,例如生物流體。流體物質可包括但不限於血液、臍帶血、唾液、尿液、汗液、血清、精液、陰道流體、胃部和消化流體、脊髓流體、胎盤流體、空腔流體、眼部流體、血清、乳房乳汁、淋巴流體或其組合。物質可為實體組織,例如生物組織。物質可包含正常的健康組織、疾病組織或健康與疾病組織的混合物。在一些情況下,物質可包含腫瘤。腫瘤可為良性的(非癌症)或惡性的(癌症)。腫瘤的非限制性實例可包括:纖維肉瘤、粘液肉瘤、脂肪肉瘤、軟骨肉瘤、骨原性肉瘤、脊索瘤、血管肉瘤、內皮肉瘤、淋巴管肉瘤、淋巴管內皮肉瘤、滑膜瘤、間皮瘤、尤因氏腫瘤(Ewing's),平滑肌肉瘤、橫紋肌肉瘤、胃腸系統癌瘤、結腸癌瘤、胰腺癌、乳房癌、泌尿生殖系統癌瘤、卵巢癌、前列腺癌、鱗狀細胞癌瘤、基底細胞癌瘤、腺癌瘤、汗腺癌瘤、皮脂腺癌瘤、乳頭狀癌瘤、乳頭狀腺癌瘤、囊腺癌瘤、髓樣癌瘤、支氣管癌瘤、腎細胞癌瘤、肝細胞瘤、膽管癌瘤、絨毛膜癌瘤、精原細胞瘤、胚胎性癌瘤、維爾姆斯氏腫瘤(Wilms'tumor)、子宮頸癌、內分泌系統癌瘤、睪丸腫瘤、肺癌瘤、小細胞肺癌瘤、非小細胞肺癌瘤、膀胱癌瘤、上皮癌瘤、神經膠質瘤、星形細胞瘤、髓母細胞瘤、顱咽管瘤、室管膜瘤、松果體瘤、成血管細胞瘤、聽神經瘤、少突神經膠質瘤、腦膜瘤、黑素瘤、成神經細胞瘤、成視網膜細胞瘤或其組合。物質可與各種類型的器官相關。器官的非限制性實例可包括腦、肝、肺、腎、前列腺、卵巢、脾、淋巴結(包括扁桃體)、甲狀腺、胰腺、心臟、骨骼肌、腸、喉、食管、胃或其組合。在一些情況下,物質包含多種細胞,包括但不限於:真核細胞、原核細胞、真菌細胞、心臟細胞、肺細胞、腎細胞、肝細胞、胰腺細胞、生殖細胞、幹細胞、誘導多能幹細胞、胃腸道細胞、血細胞、癌細胞、細菌細胞、從人微生物組樣品分離的細菌細胞等。在一些情況下,物質可包含細胞的內容物,諸如單一細胞的內容物或多個細胞的內容物。用於分析單個細胞的方法和系統提供於2014年6月26日提交的例如美國臨時專利申請號62/017,558中,該臨時專利申請的全部公開內容以全文引用的方式併入本文中。
可從各種受試者獲得樣品。受試者可為活受試者或死受試者。受試者的實例可包括但不限於人、哺乳動物、非人哺乳動物、齧齒動物、兩棲動物、爬行動物、犬、貓科動物、牛、馬、山羊、綿羊、母雞、禽類(avine)、小鼠、兔、昆蟲、蛞蝓、微生物、細菌、寄生蟲或魚。在一些情況下,受試者可為患有疾病或病症、被懷疑患有疾病或病症或處於發展疾病或病症的風險之中的患者。在一些情況下,受試者可為孕婦。在某一情況下,受試者可為正常的健康孕婦。在一些情況下,受試者可為處於懷有具有某種先天缺陷的胎兒的風險之中的孕婦。
可通過各種方法從受試者獲得樣品。舉例來說,可通過以下方式從受試者獲得樣品:到達循環系統(例如經由注射器或其他設備靜脈內或動脈內地),收集分泌的生物樣品(例如唾液、痰尿液、糞便等),手術(例如活檢)獲得生物樣品(例如手術中樣品、手術後樣品等),擦拭(例如頰拭子、口咽拭子),或移液。
CNV可與治療功效相關。舉例來說,增加的HER2基因拷貝數可增加晚期非小細胞肺癌中對吉非替尼治療的反應。參見Cappuzzo F.等(2005)J.Clin.Oncol.23:5007-5018。高EGFR基因拷貝數可預示對拉帕替尼和卡培他濱的敏感性增加。參見Fabi等(2010)J.Clin.Oncol.28:15s(2010年ASCO年會)。高EGFR基因拷貝數與對西妥昔單抗和帕尼單抗的敏感性增加相關。
拷貝數變異可與癌症患者對某些治療劑的抗性相關。舉例來說,胸苷酸合成酶的擴增可導致轉移性結腸直腸癌患者對5-氟尿嘧啶治療的抗性。參見Wang等(2002)PNAS USA,第99卷,第16156-61頁。
IX.計算機控制系統
本公開提供計算機系統,其經過編程或以其他方式配置成實現本文所提供的方法,諸如如本文所描述用於核酸測序和基因變異測定、存儲參考核酸序列、進行序列分析以及/或者比較樣品和參考核酸序列的方法。圖22中示出了此類計算機系統的實例。如圖22中所示,計算機系統2201包括中央處理單元(CPU,本文中也為「處理器」和「計算機處理器」)2205,其可為單核或多核處理器,或用於平行處理的多個處理器。計算機系統2201還包括存儲器或存儲位置2210(例如隨機存取存儲器、只讀存儲器、閃速存儲器)、電子存儲單元2215(例如硬碟)、用於與一個或多個其他系統通信的通信接口2220(例如網絡適配器)以及外圍裝置2225,諸如緩存、其他存儲器、數據存儲和/或電子顯示適配器。存儲器2210、存儲單元2215、接口2220以及外圍裝置2225通過通信總線(實線)(諸如母板)與CPU 2205通信。存儲單元2215可為用於存儲數據的數據存儲單元(或數據存儲庫)。計算機系統2201可在通信接口2220的輔助下可操作地耦合至計算機網絡(「網絡」)2230。網絡2230可為網際網路、網際網路以及/或者外聯網,或者與網際網路通信的內聯網和/或外聯網。網絡2230在一些情況下為電信和/或數據網絡。網絡2230可包括一個或多個計算機伺服器,所述一個或多個計算機伺服器可實現分布式計算,諸如雲計算。網絡2230在一些情況下在計算機系統2201的輔助下可實現對等網絡,所述對等網絡可使得耦合至計算機系統2201的裝置能夠起客戶端或伺服器的作用。
CPU 2205可執行機器可讀指令的序列,所述機器可讀指令的序列可在程序或軟體中實現。可將指令存儲在存儲位置(諸如存儲器2210)中。由CPU 2205進行的操作的實例可包括取指令、解碼、執行以及寫回。
存儲單元2215可存儲文件,諸如驅動器、文庫以及保存的程序。存儲單元2215可存儲用戶數據,例如用戶偏好和用戶程序。計算機系統2201在一些情況下可包括一個或多個額外數據存儲單元,所述一個或多個額外數據存儲單元在計算機系統2201的外部,諸如位於通過內聯網或網際網路與計算機系統2201通信的遠程伺服器上。
計算機系統2201可通過網絡2230與一個或多個遠程計算機系統通信。舉例來說,計算機系統2201可與用戶(例如操作者)的遠程計算機系統通信。遠程計算機系統的實例包括個人計算機(例如可攜式PC)、板型或平板PC(例如 iPad、 Galaxy Tab)、電話、智慧型手機(例如 iPhone、Android可實現裝置、)或個人數字助理。用戶可經由網絡2230訪問計算機系統2201。
可通過存儲於計算機系統2201的電子存儲位置上(諸如在存儲器2210或電子存儲單元2215上)的機器(例如計算機處理器)可執行的代碼來實現如本文所描述的方法。可以軟體的形式提供機器可執行或機器可讀代碼。在使用期間,可由處理器2205執行代碼。在一些情況下,可從存儲單元2215檢索代碼並且存儲於存儲器2210上,以備由處理器2205存取。在一些情況下,可排除電子存儲單元2215,並且將機器可執行指令存儲於存儲器2210上。
代碼可被預編譯並且被配置成與具有適合執行代碼的處理器的機器一起使用,或在運行期間被編譯。可在程式語言中提供代碼,可對所述程式語言加以選擇以使得代碼能夠以預編譯或當時編譯(as-compiled)方式執行。
可在編程中實現本文所提供的諸如計算機系統2201的系統和方法的多個方面。技術的各個方面可被認為是典型地呈在一類機器可讀介質上執行或在一種類型的機器可讀介質中實現的機器(或處理器)可執行代碼和/或相關數據形式的「產品」或「製品」。機器可執行代碼可存儲於電子存儲單元,諸如存儲器(例如只讀存儲器、隨機存取存儲器、閃速存儲器)或硬碟中。「存儲」類型介質可包括計算機、處理器等的任何或所有的有形存儲器,或其相關模塊,諸如各種半導體存儲器、磁帶驅動器、磁碟驅動器等,其可在軟體編程的任何時間提供非暫時存儲。軟體的全部或部分有時可通過網際網路或各種其他電信網絡進行通信。此類通信例如可實現軟體從一個計算機或處理器加載至另一者中,例如從管理伺服器或主機計算機至應用程式伺服器的計算機平臺中。因此,可承載軟體元素的另一類型的介質包括諸如通過有線和光學陸上線路網絡並經各種空中鏈路跨越本地裝置之間的物理接口使用的光波、電波以及電磁波。攜帶此類波的物理元素(諸如有線或無線鏈路、光學鏈路等)也可被視為承載軟體的介質。如本文中所用,除非限於非暫時有形「存儲」介質,否則諸如計算機或機器「可讀介質」等術語是指參與提供指令至處理器以執行的任何介質。
因此,機器可讀介質(諸如計算機可執行代碼)可採取許多形式,包括但不限於有形存儲介質、載波介質或物理傳輸介質。非易失性存儲介質包括例如光碟或磁碟,諸如圖式中所示的任何計算機中諸如可用於實現資料庫等的任何存儲裝置等。易失性存儲介質包括動態存儲器,諸如此類計算機平臺的主存儲器。有形傳輸介質包括同軸線纜;銅線以及光纖,包括包含計算機系統內的總線的線。載波傳輸介質可採取電或電磁信號或聲波或光波形式,諸如在射頻(RF)和紅外線(IR)數據通信期間產生的那些。因此,計算機可讀介質的常見形式包括例如:軟磁碟、軟盤、硬碟、磁帶、任何其他磁性介質、CD-ROM、DVD或DVD-ROM、任何其他光學介質、打孔卡紙帶、具有孔模式的任何其他物理存儲介質、RAM、ROM、PROM和EPROM、FLASH-EPROM、任何其他存儲晶片或盒、傳送數據或指令的載波、傳送此類載波的線纜或鏈路或計算機可從中讀取編程代碼和/或數據的任何其他介質。這些形式的計算機可讀介質中許多可參與將一個或多個指令的一個或多個序列運送至處理器以執行。
計算機系統2201可包括電子顯示器2235或與其通信,所述電子顯示器包括用於提供例如耦合至計算機系統2201的核酸測序儀器的輸出或讀出的用戶界面(UI)。此類讀出可包括核酸測序讀出,諸如包含給定核酸樣品的核酸鹼基的序列。還可利用此類讀出使用UI來展示分析結果。UI的實例包括但不限於圖形用戶界面(GUI)和基於網絡的用戶界面。電子顯示器2235可為計算機監視器,或電容或電阻式觸控螢幕。
實施例
實施例1:定相變體的識別
使用Blue Pippin DNA尺寸選擇系統對來自NA12878人細胞系的基因組DNA進行基於尺寸的片段分離以回收長度為約10kb的片段。然後使用微流體分配系統將經過尺寸選擇的樣品核酸與條形碼珠粒共分配於氟化油連續相內的水性微滴中(參見例如2014年4月10日提交並且出於所有目的以全文引用的方式併入本文中的美國臨時專利申請號61/977,804),其中水性微滴還包括dNTP、熱穩定性DNA聚合酶和用於在微滴內進行擴增的其他試劑以及用於使條形碼寡核苷酸從珠粒釋放的化學活化劑。對1ng的總輸入DNA和2ng的總輸入DNA重複此操作。獲得作為呈現超過700,000種不同條形碼序列的條形碼多樣性儲備文庫的子集的條形碼珠粒。含有條形碼的寡核苷酸包括額外序列組分並且具有以下一般結構:
珠粒-P5-BC-R1-N-mer
其中P5和R1分別指Illumina連接序列和讀段1引物序列,BC表示寡核苷酸的條形碼部分,並且N-mer表示用於引導模板核酸的隨機10鹼基N-mer引導序列。參見例如2014年6月26日提交的美國專利申請號14/316,383,該專利申請的全部公開內容出於所有目的以全文引用的方式併入本文中。
在珠粒溶解之後,對微滴進行熱循環以允許條形碼寡核苷酸針對各微滴內的樣品核酸的模板進行的引物延伸。這產生樣品核酸的拷貝片段,除了上文所闡述的其他所包括的序列,所述拷貝片段還包括代表起源分區的條形碼序列。
在對拷貝片段進行條形碼標記之後,將包括擴增拷貝片段的微滴的乳液破壞,並且通過額外擴增將額外的測序儀所需組分(例如用於Illumina測序儀的讀段2引物序列和P7連接序列)添加至拷貝片段,所述額外擴增將這些序列連接至拷貝片段的另一端。
然後在Illumina HiSeq系統上以10X覆蓋、20X覆蓋以及30X覆蓋對測序文庫進行測序,並且然後分析所得序列讀段和其相關條形碼序列。然後將共有共同條形碼的鄰近映射序列組裝成更大重疊群,並且識別單核苷酸多態性並且將其基於其相關條形碼和序列映射與單個起始分子相關聯,以識別定相SNP。然後基於重疊定相SNP將包括重疊定相SNP的序列組裝成定相序列數據的相位區塊或推測重疊群。將所得數據與供比較的細胞系的已知單倍型圖譜相比較。
在至少一種方法中,將一系列雜合變體中的各等位基因指派到兩種單倍型中的一者至兩者。定義了對數似然函數log P(條形碼化讀段|定相指派,變體),其返回所觀測到的讀段和條形碼數據(給定一組變體)的對數似然性以及雜合變體的定相指派(phasing assignment)。對數似然函數的形式源於關於條形碼化序列讀段數據的兩個主要觀察:(1)來自一個條形碼的讀段覆蓋單倍體基因組的一小部分,所以一個條形碼含有基因組的給定區域中的兩種單倍型的讀段的概率較小。相反地,基因組局部區域中的一個條形碼的讀段極有可能來自單一單倍型;(2)所觀測到的鹼基不同於其所源於的單倍型中的真實鹼基的概率是通過由測序儀指派的所觀測到的鹼基的Phred QV來描述。
然後報告了對於給定的條形碼化讀段和變體集合,使對數似然函數最大化的定相配置。然後通過結構化搜索程序找到了最大似然性評分單倍型配置。首先,使用集束搜索來找到相鄰變體(例如約50個變體)的較小區塊的最佳定相配置。其次,在區塊接合處上以掃描的形式測定諸多個區塊的相對定相。此時,找到了總體接近最優的定相配置並且用作進一步優化的起始點。然後將單個變體的單倍型指派倒位,以找到對定相局部改善,交換的配置之間的對數似然性的差異提供對定相指派的置信度的評估。最後,將定相配置分解成相位區塊,所述相位區塊具有高內部校正概率。然後,通過將最佳配置的對數似然性與其中當前SNP右側的所有SNP均使其單倍型指派倒位的配置相比較來測試是否在各SNP處破壞相位區塊。
下表提供針對NA 12878基因組獲得的定相度量。很明顯,從短讀段序列數據獲得極長相位區塊,從而正確識別顯著百分比的定相SNP,並且短切換誤差或長切換誤差極低。
其他實驗對許多額外樣品的SNP進行了定相,包括NA12878trio(NA12878、NA12882以及NA12877)、古吉拉特人(Gujarati)(NA20847)、墨西哥人(NA19662)以及非洲人(NA19701)細胞系樣品。實現約1MB的N50相位區塊長度,並且定相的SNP大於95%,並且切換誤差小於0.3%。相同樣品的全外顯子組測序(例如其中條形碼後進行靶向型向下拉動)顯示約90%的基因SNP定相,並且切換誤差再次小於0.3%。
實施例2:EML-4/ALK基因倒位/易位的識別
使用本文所描述的方法和過程來檢測所表徵的癌細胞系的結構變異。特定而言,NCI-H2228肺癌細胞系已知在其基因組內具有EML4-ALK融合易位。圖15中說明了與野生型相比的變異結構。如上部圖中所示,在變體結構中,EML-4基因(雖然在同一染色體上)與ALK基因相對隔開或遠離,實際上是易位的並且融合至ALK基因(參見例如Choi等,Identification of Novel Isoforms of the EML4-LK Transforming Gene in Non-Small Cell Lung Cancer,J.Cancer Res.,68:4971(2008年7月))。在易位的同時,EML4基因還是倒位的。圖II中進一步說明了易位,與野生型結構相比,其中易位引起EML-4的外顯子1-6(以黑框示出)與ALK的外顯子20-29(以白框示出)的融合,以及與EML-4的外顯子1-19融合的ALK的外顯子7-23的融合。
為了識別此變異,使用Blue系統(Sage Sciences,Inc.)對來自NCI-H2228細胞系的基因組DNA進行尺寸分離以選擇長度為約10kb的片段。
然後如上文關於實施例1所描述,將經過尺寸選擇的樣品核酸與條形碼珠粒共分配,擴增並且處理成測序文庫,除了在條形碼化之後並且在測序之前使用Agilent SureSelect外顯子組捕獲試劑盒對DNA進行雜交捕獲。然後在Illumina HiSeq系統上對測序文庫進行測序達到約80X覆蓋,並且然後分析所得序列讀段和其相關條形碼序列。與野生型相比,顯然基因組中跨越易位事件的部分之間共有的條形碼的數目更高,說明不存在於野生型中的融合組分之間的結構接近性。特定而言,並且如圖16A中所示,融合結構顯示12個條形碼在EML-4外顯子1-6與ALK外顯子20-29之間以及20個條形碼在EML-4外顯子7-23與ALK外顯子1-19之間的條形碼重疊,這類似於雜合細胞系的野生型構建體的重疊條形碼。
相比之下,如圖16B中所示,使用非變體細胞系(NA12878)進行的陰性對照運行僅大體上顯示野生型相較於變體構建體的條形碼重疊,並且序列覆蓋為約140X,並且使用3ng的起始DNA。
特定而言,雖然對各個序列區段展示較大數目的全部映射條形碼,但通過與展現極高數目的共同或重疊條形碼的野生型結構相比較僅在融合結構中看到極小百分比(例如全部映射條形碼的少於0.5%)的重疊條形碼。因此,跨融合或易位斷點的共同映射條形碼提供識別那些易位事件的強大基礎。
還採用了用於SV檢測的算法,其首先搜索具有顯著條形碼相交/重疊的所有基因組基因座對,以高效稀疏矩陣相乘對此搜索進行編碼。然後利用合併讀段對、分離讀段以及條形碼數據的概率模型對此第一階段的候選物進行過濾。NA12878和NA20847上的SV識別使得識別多個大規模缺失和倒位,並且相對於鄰近相位區塊對其進行定相,從而在上文所描述的核trio中在諸多種遺傳模式下顯示定相一致性。
實施例3:經由CNV篩檢來檢測增加的狼瘡易感性
測試患者對狼瘡的易感性。從患者抽取血液。使用本文所敘述的技術對無細胞DNA樣品進行測序。然後將序列與已知基因組參考序列相比較以測定不同基因的CNV。FCGR3B(CD16細胞表面免疫球蛋白受體)的拷貝數低指示對系統性紅斑狼瘡的易感性增加。患者被告知任何拷貝數失常和相關風險/疾病。
實施例4:經由CNV篩選來檢測增加的成神經細胞瘤傾向
測試患者的成神經細胞瘤傾向。從患者抽取血液。使用本文所敘述的技術對無細胞DNA樣品進行測序。然後將序列與已知基因組參考序列相比較以測定不同基因的CNV。在1q21.1處的CNV指示增加的成神經細胞瘤傾向。患者被告知任何拷貝數失常和相關風險/疾病。
實施例5:經由CNV篩選來鑑別診斷肺癌
對慢性咳嗽、體重減輕以及呼吸急促患者進行關於肺癌的測試。從患者抽取血液。使用本文所敘述的技術對循環腫瘤細胞(CTC)或無細胞DNA樣品進行測序。然後將CTC序列與已知基因組參考序列相比較以測定不同基因的CNV。如果DNA中的EGFR拷貝數高於正常,那麼可將患者鑑別診斷為患有非小細胞肺癌(NSCLC)而不是小細胞肺癌。非小細胞肺癌的CTC還具有其他拷貝數變異,所述其他拷貝數變異可使其與小細胞肺癌進一步區分開。視癌症的階段而定,囑咐進行手術、化學治療或放射治療。
小細胞肺癌很多時候比非小細胞肺癌瘤更快速並且廣泛地轉移(並且因此被區別地劃分階段)。NSCLC通常對化學治療和/或放射不太敏感,因此,如果被診斷處於早期,手術為所選治療,經常使用涉及順鉑的輔助(輔佐)性化學治療。非小細胞肺癌(NSCLC)患者還可用靶向治療,例如ALK抑制劑,諸如克唑替尼。靶向治療通過幹擾為致癌作用和腫瘤生長所需的特定靶向分子,而不是通過簡單地幹擾所有快速分裂的細胞(例如在傳統化學治療情況下)來阻礙癌細胞生長。
實施例6:經由定相來鑑別診斷胎兒非整倍性
胎兒非整倍性為染色體數目失常。非整倍性通常引起顯著的物理和神經損傷。X染色體數目減少引起特納氏症候群。21號染色體的拷貝數增加引起唐氏症候群。侵入性測試(諸如羊膜穿刺術或絨毛膜絨毛取樣(CVS))可導致流產風險,並且此處使用較無創的測試母體血液的方法。
對具有唐氏症候群或特納氏症候群家族史的懷孕患者進行測試。收集含有胎兒基因材料的母體血液樣品。然後如本文所描述將來自不同染色體的核酸與條形碼化標籤分子一起分離至不同分區中。然後對樣品進行測序,並且將各染色體拷貝的數目與正常二倍體染色體上的序列相比較。患者被告知不同染色體的任何拷貝數失常和相關風險/疾病。
實施例7:經由定相來檢測染色體易位以鑑別診斷伯基特氏淋巴瘤(Burkitt’s Lymphoma)
伯基特氏淋巴瘤的特徵為染色體中的t(8;14)易位。對總體上被診斷為具有淋巴瘤的患者進行關於伯基特氏淋巴瘤的測試。從淋巴結收集腫瘤活檢標本。如本文所描述將來自不同染色體的核酸與條形碼化標籤分子一起分離至不同分區中。然後對樣品進行測序,並且與對照DNA樣品相比較以檢測染色體易位。如果患者被診斷為具有伯基特氏淋巴瘤,那麼可能需要比在其他類型的淋巴瘤的情況下更強的化學治療方案,包括CHOP或R-CHOP方案。CHOP由以下組成:環磷醯胺,一種烷基化劑,其通過與DNA結合併且使得形成交聯來損害DNA;羥基佐柔比星(Hydroxydaunorubicin)(也稱為多柔比星或阿黴素(Adriamycin)),一種嵌入劑,其通過將自身插入DNA鹼基之間來損害DNA;安可平(Oncovin)(長春新鹼(vincristine)),其通過與微管蛋白結合來阻止細胞複製;潑尼松(Prednisone)或潑尼松龍(prednisolone),其為皮質類固醇。還可將此方案與單克隆抗體利妥昔單抗組合,因為伯基特氏淋巴瘤是來源於B細胞;此組合被稱為R-CHOP。
實施例8:通過與親代基因組比較來定相源於無細胞DNA的胎兒基因組序列
收集來自懷孕患者的包含母體DNA的樣品和來自胎兒父親的包含父體DNA的樣品。如本文所描述將來自各樣品的核酸與分子條形碼化標籤一起分離至不同分區中。然後對樣品進行測序,並且使用所述序列來產生所分配的母體和父體片段中的每一者的推測重疊群。使用推測重疊群來構建母體和父體染色體中的每一者的諸多個部分的單倍型區塊。
收集含有胎兒基因材料的母體血液樣品。對無細胞DNA進行測序以產生母體循環DNA與胎兒循環DNA的序列。將讀段與上面所產生的父體和母體相位區塊相比較。一些相位區塊在減數分裂期間已經歷重組。識別與父體相位區塊而不與母體相位區塊匹配的胎兒材料。在一些情況下,胎兒材料與整個父體相位區塊匹配,並且確定胎兒具有父體遺傳染色體中的所述父體相位區塊。在其他情況下,胎兒材料與一個相位區塊的一部分匹配,並且然後與第二相位區塊匹配,其中這兩個相位區塊均位於父體基因組中的同源染色體區域上。確定在此區域發生減數分裂重組事件,確定最有可能的重組點,並且產生作為兩個父體相位區塊的組合的新穎胎兒相位區塊。
將循環DNA的序列與母體相位區塊相比較。使用母體相位區塊中的雜合性位點來確定源於母體的胎兒染色體的最有可能的相位。使用循環DNA序列來測定母體基因組的雜合位點處的拷貝數。特定母體相位區塊的拷貝數升高指示胎兒中源於母體的染色體含有升高的相位區塊的序列。在一些情況下,類似於在父體情況下所描述,首先同源區域的一個相位區塊將出現升高,並且然後同一區域的另一相位區塊的一部分將出現升高,表明減數分裂重組已發生。在這些情況下,確定最有可能的重組區域,並且由兩個母體相位區塊構建新的胎兒相位區塊。
雖然本文中已示出和描述了本發明的優選實施方案,但對本領域技術人員來說將顯而易見的是此類實施方案僅僅是通過舉例而提供。本發明不旨在受說明書內所提供的特定實施例限制。雖然已參照上述說明書描述了本發明,但本文中對實施方案的描述和說明並不意在以限制意義來解釋。本領域技術人員將會想到許多變化、改變以及替換,而不會脫離本發明。此外,應了解,本發明的所有方面不限於本文所闡述的特定描述、配置或相對比例,其取決於多種條件和變量。應了解,在實踐本發明時可採用本文中所描述的本發明實施方案的各種替代方案。因此可以預期的是,本發明還應涵蓋任何此類替代、修改、變化或等效物。以下權利要求旨在限定本發明的範圍並且從而涵蓋這些權利要求和其等效物的範圍內的方法和結構。